R 报错 Error protect() protection stack overflow
用Rstudio跑程序的时候报错:
|
|
大部分解决方案是在代码中设置options(expressions = 5e5),但不能解决。其实是在执行R代码是遇到防护堆叠上溢的error,但实际上服务器的内存很大,我们增加指针保护堆栈大小就行,但是要先把R程序准备成脚本,用Rscript运行的时候添加–max-ppsize选项。例如
|
|
顺便学习下R的内存控制,mark一下

用Rstudio跑程序的时候报错:
|
|
大部分解决方案是在代码中设置options(expressions = 5e5),但不能解决。其实是在执行R代码是遇到防护堆叠上溢的error,但实际上服务器的内存很大,我们增加指针保护堆栈大小就行,但是要先把R程序准备成脚本,用Rscript运行的时候添加–max-ppsize选项。例如
|
|
顺便学习下R的内存控制,mark一下
exceRpt:The extra-cellular RNA processing toolkit
关于miRNA的定量工具其实有很多,有人用STAR或者bowtie比对,然后自己定量。我喜欢打包的解决方案,避免造轮子,我经常用的是miRDeep2。朋友问piRNAR如何定量,是否可以用exceRpt。于是和他一起研究了下exceRpt,发现exceRpt对miRNA的定量基本上与miRDeep2的结果一致,感觉exceRpt靠谱。
exceRpt目地址:http://github.gersteinlab.org/exceRpt/
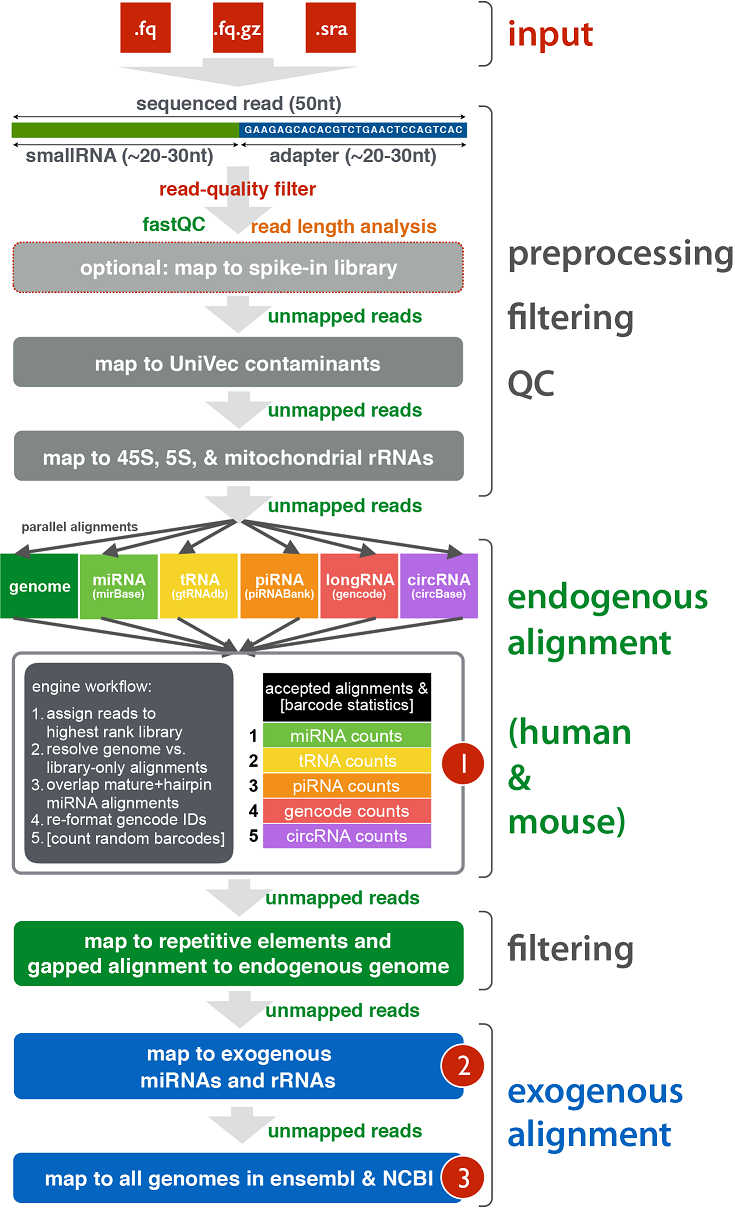
主要思想是先去掉无关的reads,比如45S, 5S,rRNA等,然后同时比对genome, miRNA(mirBase),tRNA( gtRNAdb), piRNA( piRNABank),longRNA(gencode), circRNA(circBase),看reads和哪种类型的的RNA最接近,然后定量。
总结连锁不平衡的一些知识,内容来源(copy)网络。
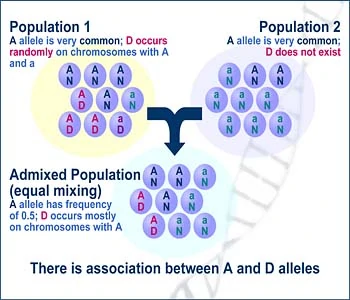
连锁不平衡(linkage disequilibrium, LD)分析是群体遗传学研究中常见的分析内容,当位于某一座位的特定等位基因与另一座位的某一等位基因同时出现的概率大于群体中因随机分布的两个等位基因同时出现的概率时,称这两个座位处于连锁不平衡状态。
连锁不平衡性是指在两个或者多个位点上的非随机关联性,这些位点既可能在同一条染色体上,也可以在不同的染色体上。连锁不平衡性也被称作配子水平的不平衡性或配子不平衡性。从另一个角度讲,连锁不平衡是等位基因或者遗传标记在一个人群中表现出高于或低于由等位基因的随机频率而预测的单模标本的频率。连锁是指染色体上的两个或者多个位点进行有限的组合,而连锁不平衡性不等同于连锁。连锁不平衡的数量取决于观察和预期的位点频率的差异。对于那些重组后位点或者基因型的频率等于预期的群体我们称其为连锁平衡。 连锁不平衡的程度取决于多方面的因素,包括遗传连锁,选择,和重组的概率,遗传漂变,选型交配以及群体结构,都会影响LD的变化。
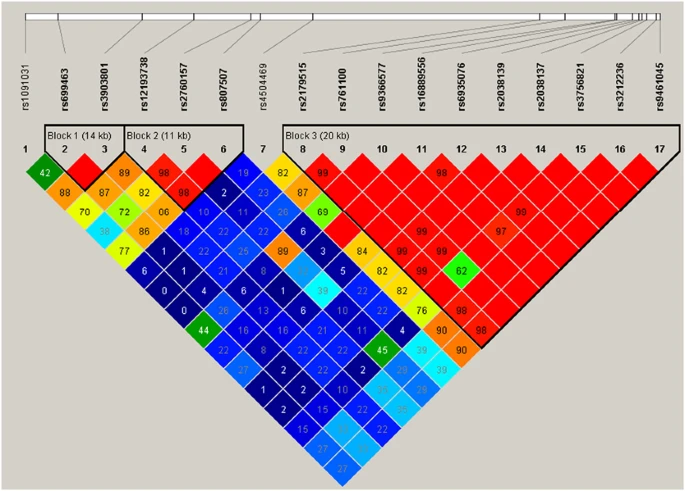
上图展示了由Haploview软件生成的连锁不平衡块Linkage disequilibrium (LD) block,LD用 D′ statistic衡量,颜色从蓝到红表示LD的高低(https://doi.org/10.1038/jhg.2016.40)。
用R画图的时候,报错,但其实代码是没问题的
1 2 3 4 5 6Error in grid.Call(C_convert, x, as.integer(whatfrom), as.integer(whatto), : VECTOR_ELT() can only be applied to a 'list', not a 'NULL' Error in grid.newpage() : could not open file '/tmp/RtmpCWAOi6/5413447bbbfc43748734b70eb6a8f054.png' Error in file(out, "wt") : cannot open the connection
升级fs包就行了。
|
|
可能升级fs包适合我的例子。如果你的问题是下面的,你可能仅仅需要拉大Rstudio显示图片的面板,重新画图即可
|
|