Convolutional Neural Networks
保存几张1D卷积的图和文章,方便以后查找,摘自互联网。说不准以后搞深度学呢🫠🙃卷的结果就是以后不上人工智能都不要意思说自己是搞生信的。
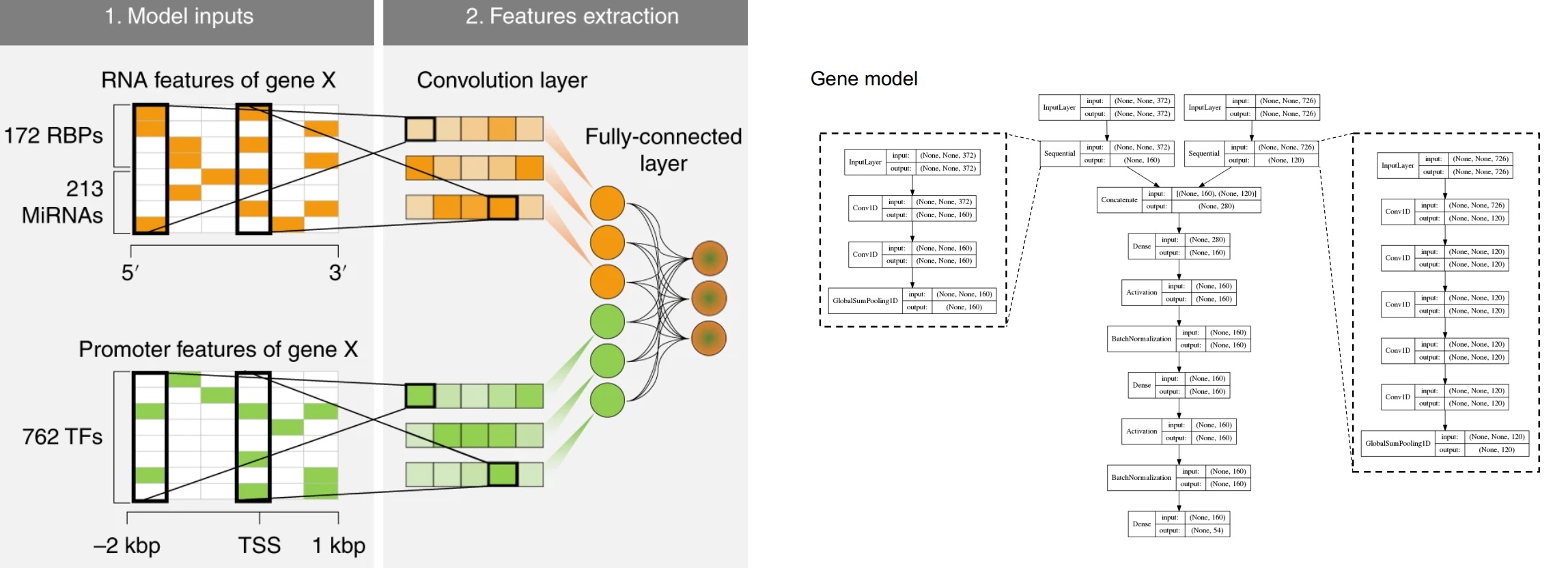
主要是看到一篇文章,用了卷积神经网络,我一直就好奇组学数据怎么做卷积,所以就看了下文章,发现用的是Conv1D。(不知道理解的对不对)对于1D卷积Conv1D而言,如果卷积核kernel size是2的话,最终会生成一个 “行数-kernel size+1“的向量,如果数据分批给的话,就有batch,比如如果样本是21000,batch size是128的话,每个batch有165个样本,所以Nature Machine Intelligence的附图 Fig. 10这篇文章还进行了BatchNormalization。
Samples = 21000,batch_size=128-> training_sample for each epoch =21000/128 = 164.06 ~= 165
filters可以指定多次卷积(相同kernel size),这样可以生成二维的数据。
| Args | https://www.tensorflow.org/api_docs/python/tf/keras/layers/Conv1D |
|---|---|
filters |
Integer, the dimensionality of the output space (i.e. the number of output filters in the convolution). |
kernel_size |
An integer or tuple/list of a single integer, specifying the length of the 1D convolution window. |
Deep learning decodes the principles of differential gene expression