通路富集分析计算显著性
富集分析 在组学分析中,会得到一组特定意义的基因集,比如差异表达基因集,然后这些基因分布在哪些通路上,是随机分布在各个通路上还是富集在了某个通

官网提供的推荐流程错误非常多(年代久远??),迄今还没有人详细介绍正确的Varscan copynumber Recommended Workflow。本文改正了官网的recommend workflow,提供正确的pipeline供大家一起学习(我相信我是第一个提供完整流程的哈)。本流程的搭建和解决方案来自网络搜索,感谢万能的网络。本文 没有对命令和输出格式做过多说明,请参阅官方文档。
varscan可以通过配对的肿瘤和组织样本,看覆盖到同一区域内的reads在肿瘤和组织样本中的差异,来检测肿瘤组织中的CNV。
Varscan提供的命令
java -jar varscan.jar copynumber $normal.pileup $tumor.pileup out 或 java -jar varscan.jar copynumber $normal-tumor.mpileup --output-file out
结果如下:
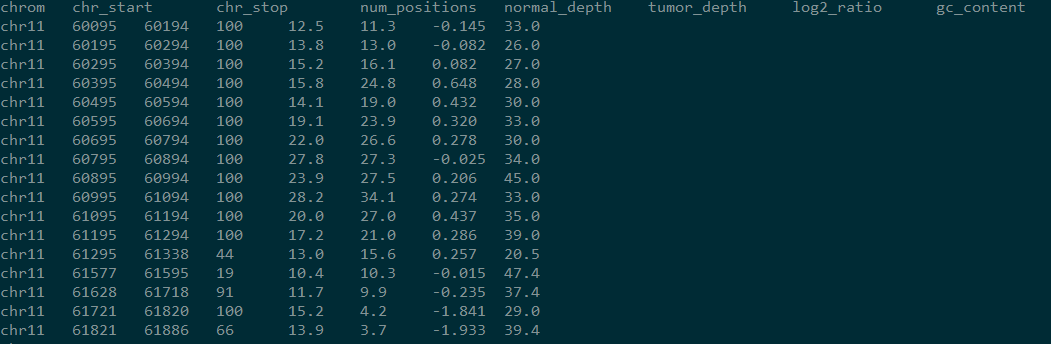
但是该结果只说明了某段区域内,肿瘤和组织depth的差异,区域相连,并没有指明哪个区域是CNV区域,发生了deletion还是insertion等。于是varscan又提供了Recommended Workflow,据说是别人提供给varscan作者的,bug百出惨目忍住。本文会在下文提供正确的流程。在此先提下Recommended Workflow的原理。
都知道在高通量测序之前,用的是生物芯片。芯片上的每个点都是基因组上的一个marker,通过检测肿瘤样本和组织样本中同一marker荧光强度比值,找到染色体上比值发生改变的位点,然后推荐CNV区域。其中有一种算法,叫做circular binary segment CBS环状二元分割算法(恕我愚笨,不了解这种算法)。R语言包DNAcopy利用CBS和每个marker的lg2ratio,判断那些区域是CNV。那高通量测序varscan的结果如何利用CBS算法呢。从varscan的输出结果可以看出,varscan提供了一定区域内的lg2ratio,Recommended Workflow就将这个区域的起始位点当作这个区域的marker,并与该区域的lg2ratio对应,于是便和芯片检测CNV的方法对接上去了。
Annovar的注释结果,如果输出制表符分割的VCF格式,显得混乱。如果输出为csv格式,方便windows下的用户用excel打开,但不方便数据处理,比如某一列的注释信息中包含了逗号,这种情况就要特别注意。python中有csv模块可以方便的读取csv,推荐使用。
本文写的小脚本主要处理简单的csv格式,亮点在于支持接收标准输入和标准输出,方便生物信息多命令之间通过管道进行处理。如果没有指定输入文件,则读取管道流数据,如果没有指定输出文件,则可以用管道接收数据进行下一步处理。
比如 cat xxx.csv ' python convert.py ' grep "xx" > result.txt
或者 python convert.py -i input.csv ' grep "xx" > result.txt
或者 python convert.py -i input.csv - o result.txt
查看用法 python convert.py --help
statistic GC content by interval. BED format file include arget interval information. BEDTool statistic read number and extract sequence, awk statistic GC content.
bedtools map -a interval.bed -b sample.bam -c 10,10 -o count,concat | awk -v OFS="t" '{n=length($5); gc=gsub("[gcGC]", "", $5); print $1,$2,$3,$4,gc/n}'
思路:利用bedtools的map工具,首先找到map到interval.bed中的每个interval的reads的序列,然后统计这些序列中有多少GC。