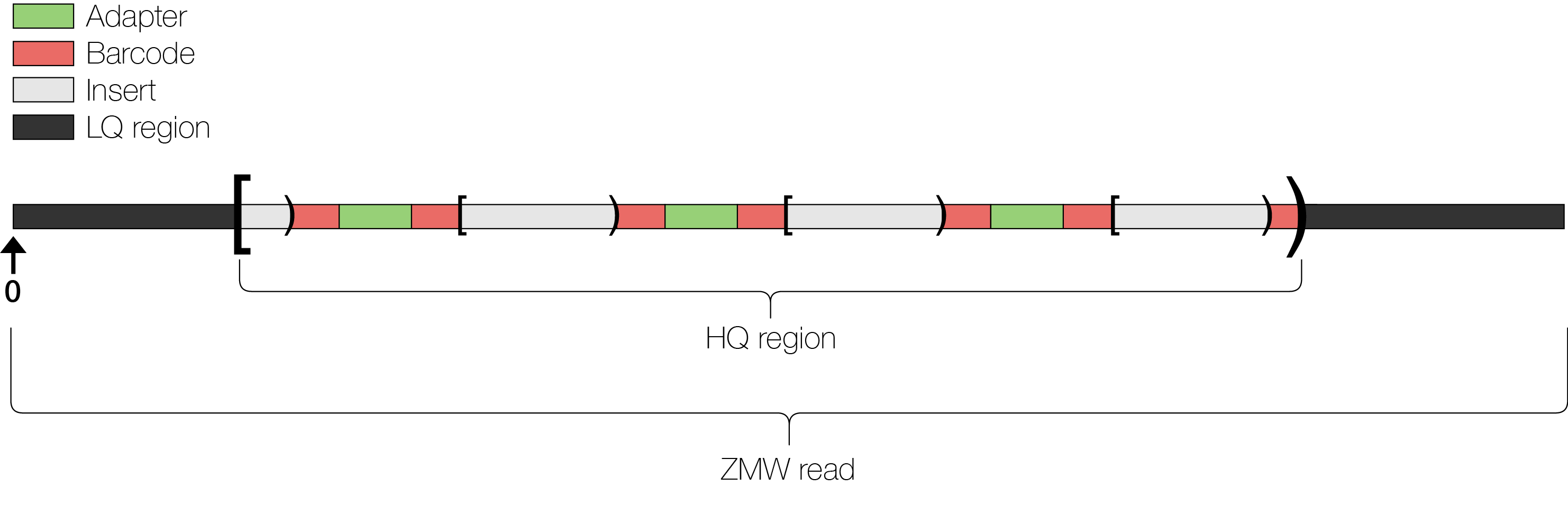
T细胞,B细胞,抗原,CD4和CD8
T细胞工作原理
T细胞在胸腺中发育后,它们可以在血液或淋巴系统中游走或迁移到体内的不同器官。只要特定的入侵者刺激它们,辅助性T细胞就会产生化学物质。有些化学物质触发B细胞发育成浆细胞,而另一些化学物质则刺激杀伤性T细胞靶向并杀死可能被侵入者感染或癌变的细胞。 调节性T细胞有助于控制免疫反应,防止其失控。自然杀伤T细胞也产生化学物质,以帮助调节免疫反应,防止入侵者和肿瘤。在免疫反应结束后,记忆T细胞在体内停留很长一段时间。这样,如果同样的入侵者再次出现,它们就能迅速做出反应,并繁殖产生大量的T细胞来消灭它。