统计GTF文件中转录本的长度 Calculate transcript length from gtf file
gtf 文件INPUT
|
|

gtf 文件INPUT
|
|
注释有很多版本,比如ensembl,gencode, ucsc known gene, NCBI的RefSeqGene。最近就需要NM id的注释,但NCBI提供的是gff3格式的,而且很乱。用UCSC table browser下载的gtf版本的RefSeq,没有转录本和基因之间的关系,也没有基因symbol。
比如Ensembl,其实Ensembl的gtf挺好用的,不过这次我因为需要NM编号的注释(笨方法是将ensembl id转成NCBI的refSeq的ID,但这不是最优的方法,ID mapping有可能对不上,不如直接用NM的注释)。
|
|
UCSC table browser下载的refGene的gtf,这个文件不对的地方是gene id和transcript id是一个,而我需要gene和transcript的关系
|
|
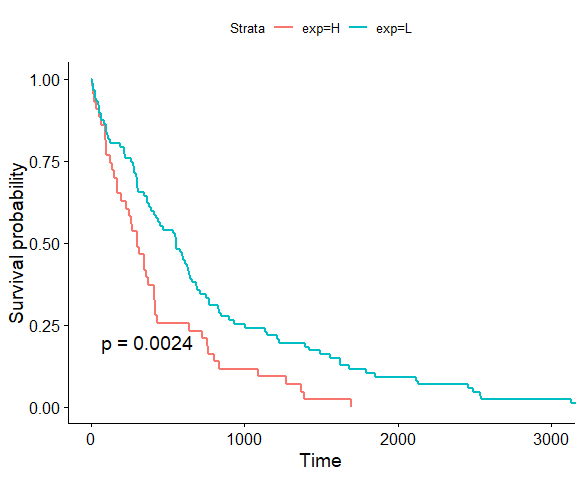
以前的工作是全基因组或全外分析,不涉及癌症和生存分析,但现在的工作主要围绕癌症方面,生存分析一定少不了。实验室小伙伴推荐用TCGAbiolinks下载TCGA的数据,于是研究了如何用TCGABiolinks下载TCGA的数据,以下载RNA的count数据为例,并做生存分析。
因为对hyper-v不是很熟悉,点了一下检查点,生成了一个avhdx文件,这个文件其实后续hyper-v会将其合并到vhdx的虚拟磁盘中。而我当时手贱手工的删除了avhdx文件,导致hyper-v找不到这个文件,vhdx也挂起等待合并,虚拟机迟迟不能启动。
有一种解决办法是文件恢复,但我用了几个文件都没有恢复成。实验室师兄(超级牛)新建了一个虚拟机挂载已有的vhdx文件,尝试用vhdx文件启动,显示不能启动,但在新的虚拟机下没有提示要合并,提示老系统的vhdx还有戏。
于是又新建了一个虚拟机实例,创建虚拟机实例之后,尝试将以前的vhdx文件挂载到新的虚拟机上,重启发现竟然以老的系统启动了。感谢能够启动,避免实验室的数据丢失。
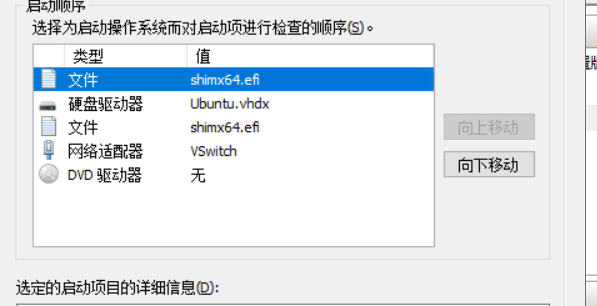
根据结果反推,第一个shimx64.efi和Ubuntu.vhdx都是以前的系统,第二个shimx64.efi新的虚拟机的,硬盘驱动器已经换成了老系统的。
STAR-FUSION和FusonAnnotator都属于Trinity Trinity Cancer Transcriptome Analysis Toolkit Fusion-finding modules。 CTAT_HumanFusionLib现阶段整合了各种资源帮助分析癌症生物学相关的fusion,同样也鉴别可能在正常样本只能出现的fusion。下载地址:https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/
FusionAnnotator –genome_lib_dir GRCh37_gencode_v19_CTAT_lib_July192017/ctat_genome_lib_build_dir/
–annotate fusions.list.txt
fusions.list.txt为star-fusion的结果中的第一列,两个参与融合的基因中间用–连在一起,就可以用FusionAnnotator进行注释,相关的标签会注释到融合基因上。
会有三类标签,每类下面又有很多具体的来源标签: Fusions relevant to cancer biology Individual genes of cancer relevance, which may show up in fusions Red Herrings: Fusion pairs that may not be relevant to cancer, and potential false positives.
通过注释,就可以了解到分析结果中的融合基因是否在其他数据库中出现过,或者可能是和癌症无关的突变。