nothing provides icu 54.* needed by r-base-*
Problem: nothing provides icu 54.* needed by r-base-**
https://github.com/rrwick/Trycycler/issues/13
1 conda config --add channels conda-forge

公开的转录组数据集多为原始形式,格式不统一、处理方式各异,难以有效利用,所以我想找一个经过标准流程清洗了GEO/SRA数据集的数据库,这样可以直接用清洗后的结果,而不用每个数据集单独处理。recount2(https://jhubiostatistics.shinyapps.io/recount/)就是类似的数据库,但只提供了2038个数据集,远远不够。
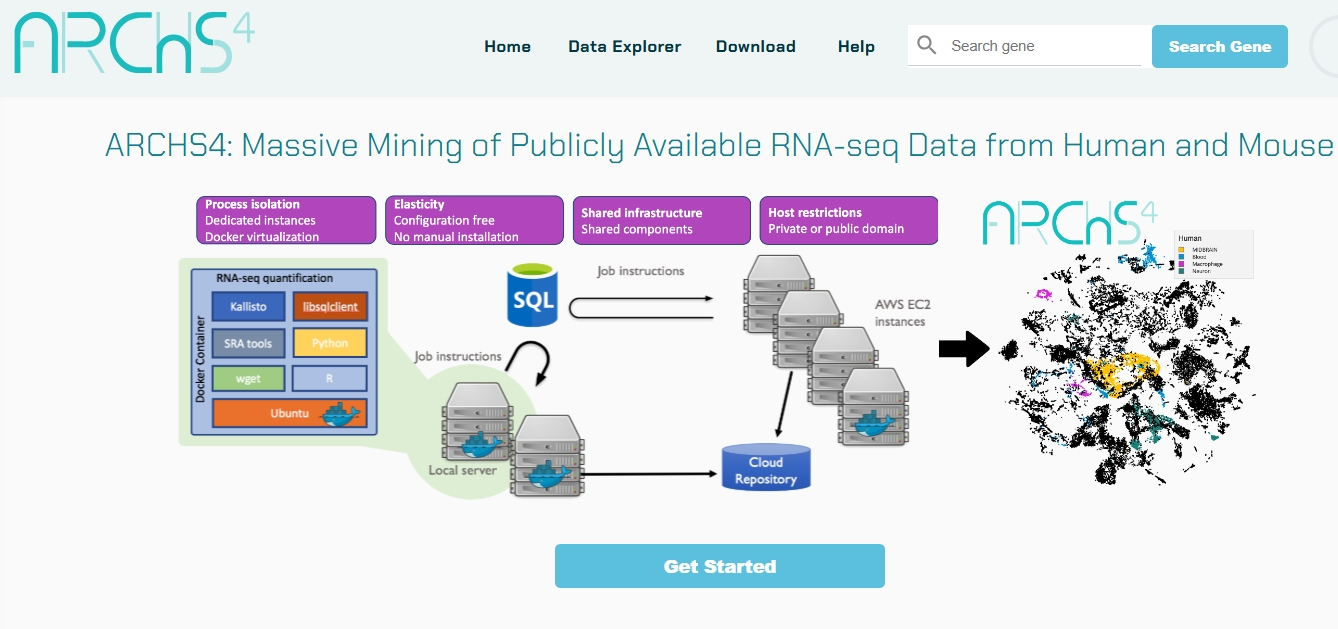
今天发现了ARCHS4(https://archs4.org/),感觉满足我目前的需求。ARCHS4数据源于GEO 和 SRA 数据库,还纳入了线虫、果蝇等其他物种的 35000 个样本数据,用的比对软件是Kallisto。数据维护一直在更新,还提供了分析和下载工具(https://github.com/MaayanLab/archs4py)。
RNA-Seq这块,提供了966693个样本比对结果,还提供了样本的tSNE结果,基因相关性结果。
对于热图大家都熟悉,热图中的颜色表示值的大小。但前几天看到一个热图,里面的颜色含有两个维度的信息,专门有一个颜色的图注(非主图,而是热图里面的颜色说明),如下,横坐标表示D值的大小,纵坐标表示的P值,可以看出二维区域中不同位置的颜色,同时反应了D值和P值的信息。

找了很多工具和方法,都不太方便实现,或者不太容易控制颜色的分布。后来突然悟了,我可以自己生成这个图,从左到右反应的D值大小,两个颜色渐变,从上往下反应的是P值显著性,颜色越来越深。
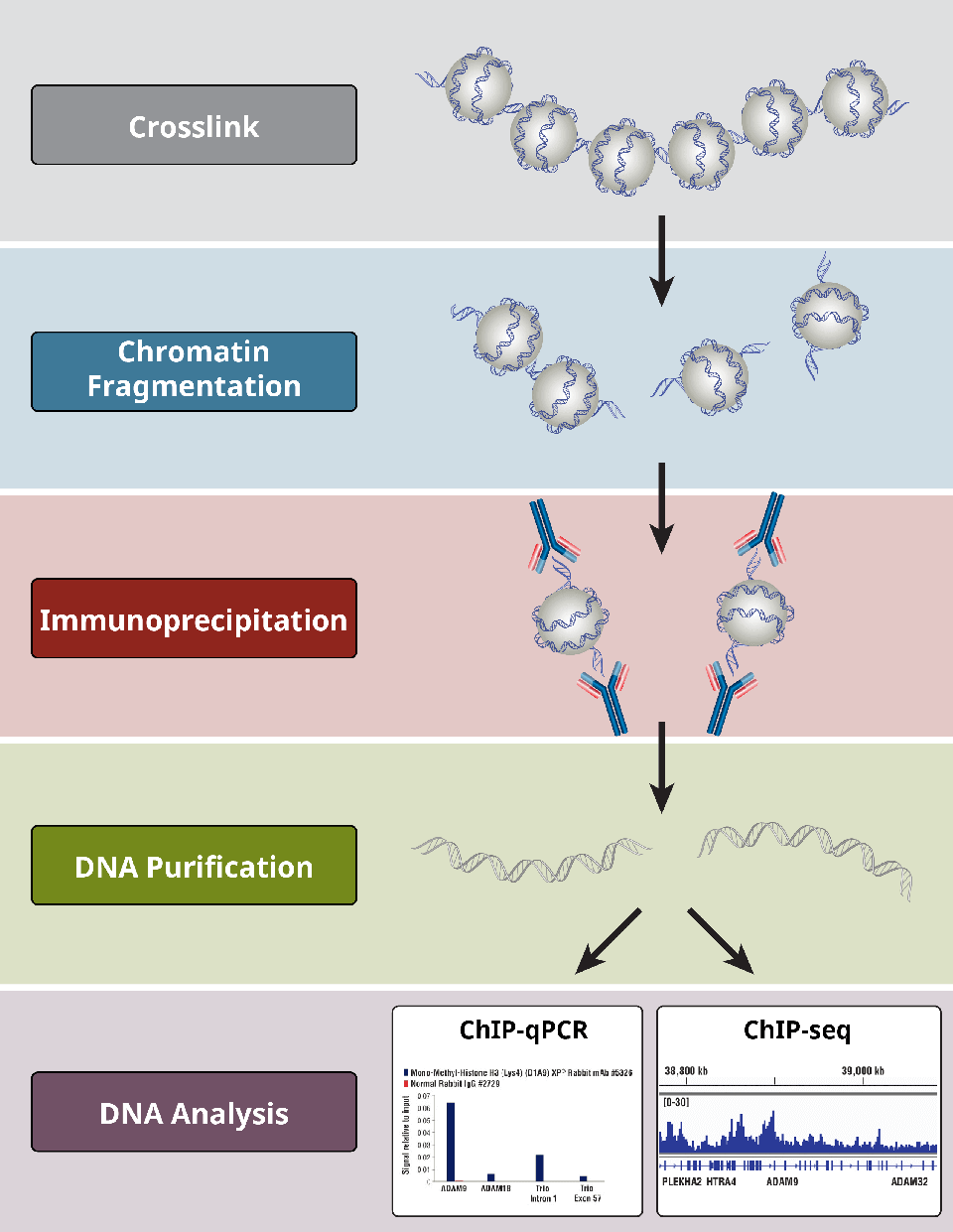
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP)与二代测序相结合的表观遗传研究技术,能够高效地在全基因组范围内对DNA和蛋白的相互作用进行检测,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。
染色质免疫沉淀法 (ChIP) 是一种基于抗体的技术,可用来选择性地使特异性 DNA 结合蛋白及其 DNA 靶标富集。ChIP 可用来研究某种特殊的蛋白-DNA 相互作用、多种蛋白-DNA 相互作用或全基因组或部分基因内的相互作用。
ChIP 使用可选择性地检测和结合蛋白的抗体,包括组蛋白、组蛋白修饰、转录因子、辅因子,以提供有关染色质状态和基因转录的信息。在 ChIP 中结合使用蛋白质组分析和分子生物学技术,能够让研究者理解目的细胞或组织中的基因表达和调节。
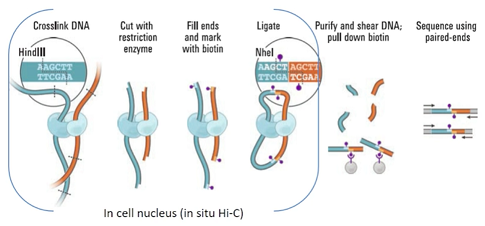
Hi-C技术源于染色体构象捕获(Chromosome Conformation Capture, 3C)技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。Hi-C技术不仅可以研究染色体片段之间的相互作用,建立基因组折叠模型,还可以应用于基因组组装、单体型图谱构建、辅助宏基因组组装等,并可以与RNA-Seq、ChIP-Seq等数据进行联合分析,从基因调控网络和表观遗传网络来阐述生物体性状形成的相关机制。