
利用biomaRt包下载HGMD公开版的突变位点
前面文章介绍了Ensembl的biomart,相信你对biomart应该有所了解了,在此再介绍一种方法,即通过R语言包biomaRt下载HGMD的数据。
HGMD的最新数据是需要购买授权才行,公开版信息不仅滞后,而且不能下载,不能得到基因组位置,在biostart上看到有人说Ensembl整合了HGMD的公开版,心想能获得公开版的数据也不错,于是采用biomaRt包下载。
各位不要高兴,最终的结果是,只得到了所有突变的基因组位置,未能下到具体的突变类型,以及与表型的关系。不过能下载基因组的位置,也算不错,结合对这些位置的注释,能获取不少信息。如果您对这些位置的利用有更多或者更好的想法,欢迎与我讨论。
1,安装biomaRt包
|
|
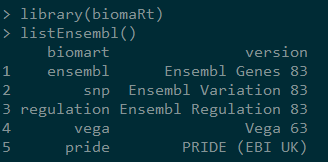
2,显示ensembl的biomart
|
|
 )
)