修改MySQL远程访问权限
修改MySQL远程访问权限
|
|
# . 表示允许所有表 ‘root’ 表示允许root用户 ‘%’ 表示任意ip ‘123456’ 表示密码
|
|

|
|
# . 表示允许所有表 ‘root’ 表示允许root用户 ‘%’ 表示任意ip ‘123456’ 表示密码
|
|
bismark调用bowtie2进行比对,调用samtools生成bam文件,因此在运行bismark之前,需要安装bowtie2和samtools
请注意,fastq文件要进行质控,比如去掉低质量的reads,去掉adaptor等,可以看本文最下方推荐的PPT,本文不介绍,此外本本只介绍到序列比对,后续的统计分析没有介绍,有兴趣的朋友可以关注swDMR和methykit工具包。
|
|
# --path\_to\_bowtie后面跟的是文件夹
# --verbose 输出log信息
# ./ref 文件夹中有一个基因组fasta文件
# --bowtie2指明用的是bowtie2
./bismark\_v0.16.1/bismark\_genome\_preparation --path\_to_bowtie /home/zzx/bowtie2-2.2.9/ --bowtie2 --verbose ./ref/
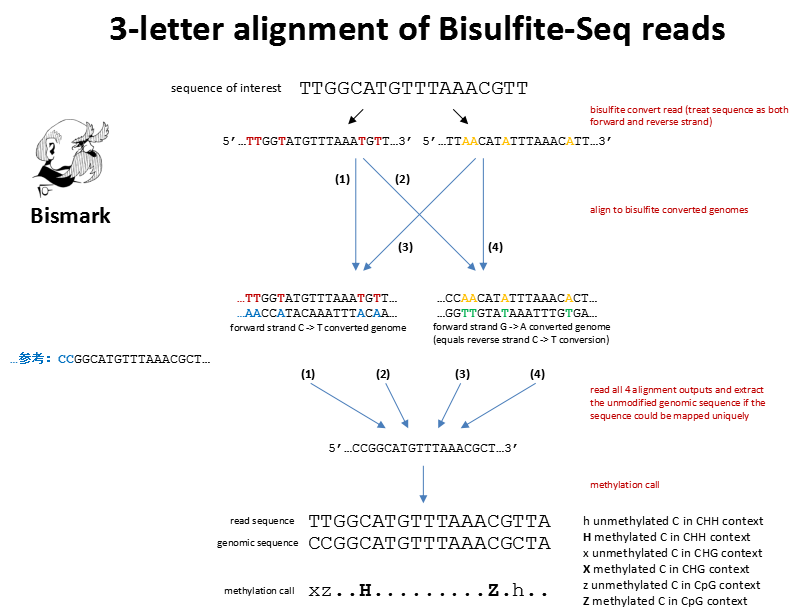
因为在重亚硫酸盐甲基化测序中,因为未甲基化的C会变为T,在正链表现为C–>T,但是在负链有C变为T,转换为正链时,即为G–>A,所以基因组需要进行两种转化,才能用于比对。在基因组目录下产生Bisulfite_Genome目录,有CT_conversion和GA_conversion文件夹,这两个文件夹包含转换后的fasta文件和bowtie2建立的索引bt2文件。
fastq中的BS转换后的read与转换的参考基因组比对,得到在参考基因组中的位置,再与原始的参考基因组比较,确定methylate call
|
|
sample_trimmed_unpaired_1.fastq.gz是sample_trimmed_unpaired_2.fastq.gz的十多倍,异常的大。1文件(forward reads)中unpaired reads非常多,显著多余2文件(reverse reads)中的unpaired reads。
awk可以通过$1,$2等,对数据进行逻辑判断或处理,比如
|
|
但如果碰到带有百分号%的数据时,则不能直接通过加减乘除进行计算或者判断
|
|
因为系统没有将1%转成数值0.01,无法进行判断$1是否等于0.01