VCF文件中的原始突变过滤--filter raw variants in vcf
Hard filter突变的传统过滤方式
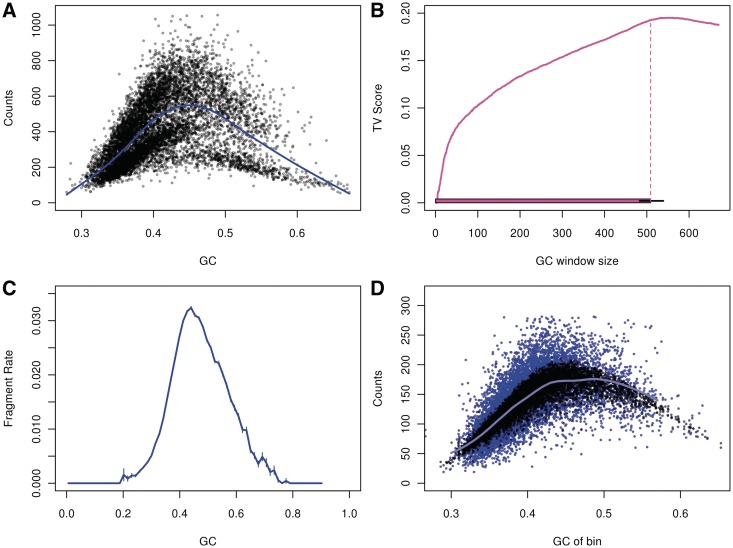
此时VCF文件中的突变,与刚开始下机得到的FASTQ文件类似,称为raw data。此时的突变集合中,有很多假阳性突变,这些突变需要在突变分析之前过滤掉。
传统的过滤方式,直接根据每个突变的注释信息,进行过滤。最直接和最常见的是根据DP标签过滤,即根据该突变位点的测序深度进行过滤。通常,深度越低,支持该突变的reads数目越少,该突变越不可信。还可以根据前面提到的QUAL质量分值进行过滤,分值越低越不可信。Forward reads和Reverse reads的比例。通过,设定一定的阈值,看这些注释信息是高于还是低于该阈值。