Parse gtf
I always use gtf file and retrieve gene information. There isn’t a highly flexible tool to solve my demand. I modified the code from “https://github.com/Jverma/GFF-Parser”, thanks Jverma. This tool will be easier to use.
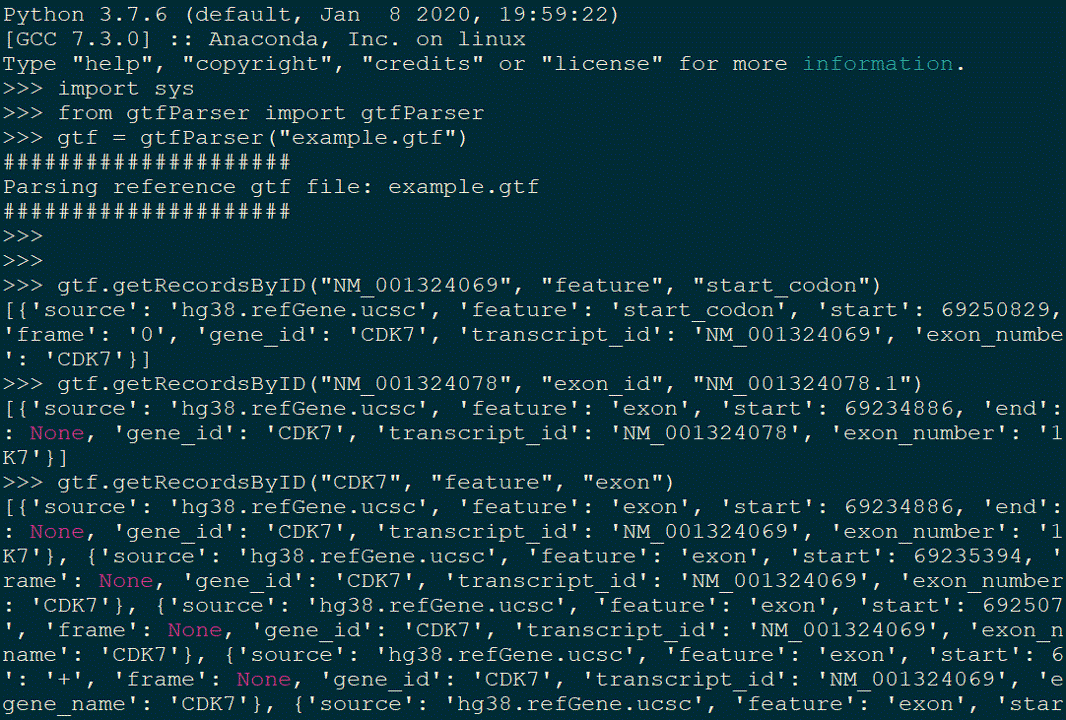

I always use gtf file and retrieve gene information. There isn’t a highly flexible tool to solve my demand. I modified the code from “https://github.com/Jverma/GFF-Parser”, thanks Jverma. This tool will be easier to use.
我是在这篇文章(Integrative pathway enrichment analysis of multivariate omics data )中遇到的合并多个p-value的操作。这篇文章是今年发表在NC上。所有的组学或者大规模的数据分析,都需要探索数据背后相关的生物学功能,所以通路富集分析非常普遍。通常的做法是基于单一组学、单一数据集的数据进行分析,随着生物学数据的爆发,大规模多组学数据变得普遍,这篇文章介绍了基于整合的多组学或多数据集的数据进行通路分析的工具ActivePathways。

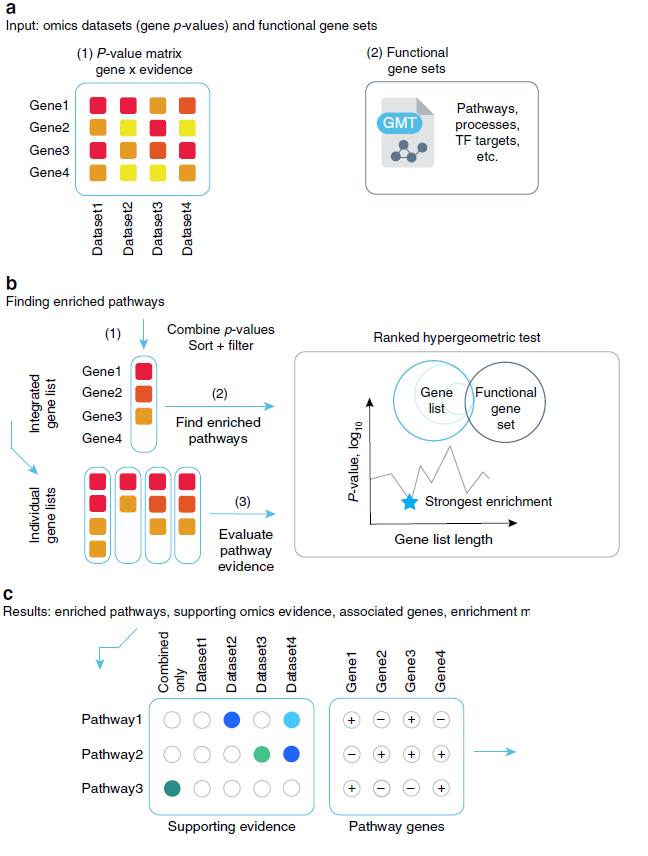
ActivePathways的方法,如下图:
(a) 需要的输入文件
(1) 基于多组学数据集的基因P-value,传统的富集分析是单组学,只有一列,现在是多组学,对应多列P-value (2) 基因集,这个和其他的通路富集分析一样,用来表示生物学过程和通路
(b)
(1) 用Brown method合并基因的P-values,并且排序,用一个宽松的阈值来过滤检阳性的基因。 (2) 对每个通路,用排序的基因(从第一个开始从少到多作为sub-list)进行超几何检验,并找到最优的sub-list长度。 (3) 基于单一组学的数据进行富集分析,找到支持每个通路的证据。
(c) ActivePathways 提供整合之后的富集分析结果,相关的Brown P-value,支持通路的证据。还可以在Cytoscape中画Enrichmentmap的图,来分析更广泛的生物学主题。点为通路,边表示有共有基因。
今天在看文章的时候,发现原来p-value也可以合并。比如一个基因在不同组学数据的检验中对应了多个p-value,可以合并成一个。
常用的是Fisher’s method,

-2[ln(P1) + ln(P2) + … + ln(Pi)]符合X2分布(自由度为2k,k为p-value的个数)。
还有Brown’s methods和 Kost’s methods,具体的介绍如下图。

M值和B值的计算公式
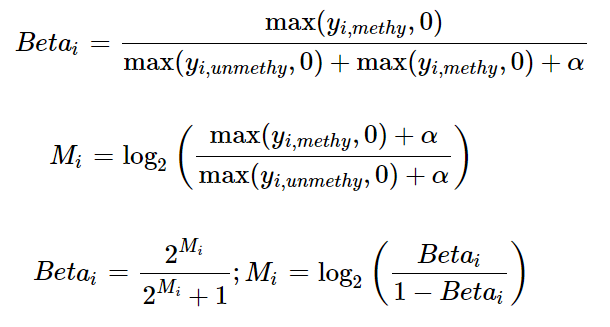
https://link.springer.com/article/10.1186/s41241-017-0041-9
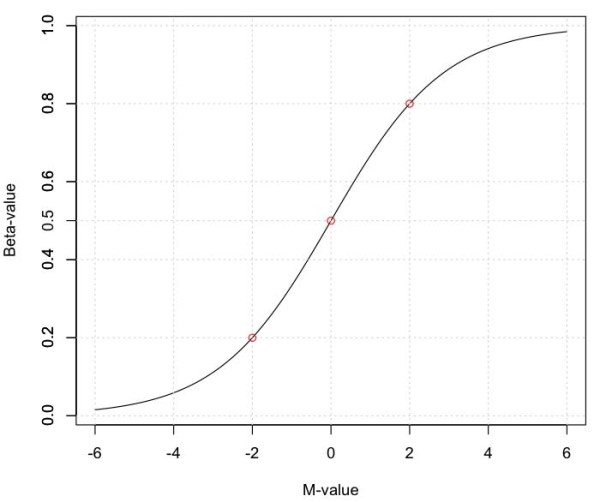
M值和B值的对应关系
我们的存储服务器有两组RAID,容量均大于150T,我在mount的时候,提示我
|
|
是因为没有分区导致的,分区之后就可以了。分区的命令
|
|