Continuous Long Read, subreads and scraps in Pacbio Raw data
Pacbio的工具更新实在是太快了,https://github.com/PacificBiosciences/pbbioconda 原来Is

帅旸谈一谈在变异解读过程中用到的几个不太熟悉的预测指标:
z score:这个指标指的是某个基因对missense的耐受程度,具体是指该基因所期望的missense数比上观察
到的missense数,如果z score>3.09,则认为该基因对missense不耐受,根据公式我们可以看出如果比值越大,则基因对missense越不耐受。利用z score可以在我们使用ACMG指南PP2的时候使用。
REVEL score:ClinGen SVI建议使用REVEL用来预测missense致病性。与其他常用missense致病性预测软件不同,REVEL整合了包括SIFT、PolyPhen、GERP++在内的13个软件的预测结果,对罕见变异的预测结果更加出色。当REVEL score>0.75,<0.15时分别使用ACMG指南PP3和BP4。
GERP++ rejected substitutions” (RS) score:GERP++从基因进化速率角度预测位点保守性,具体是指该基因位点所期望的碱基替换次数减去观察到的碱基替换次数,可见分数越大,该位点保守性较强,当GERP++ RS score>6.8时,认为该位点保守。当分析一个不影响剪切的同义突变时,如果RS score<6.8,则可以使用ACMG指南BP7。
dbscSNV score:dbscSNV含有两个不同的算法,用来预测变异是否影响截切,一个是基于adaptive boostin,一个是基于Random Forest。当两种算法得分均小于0.6时,则认为不影响剪切。
https://github.com/MilesMcBain/datapasta/
还在手工的把excel的数据写成导到R里吗。不管横着还是竖着复制数据,datapasta可以自动、快速的把复制数据转成tibbles, data.frames, 或者 vectors格式。
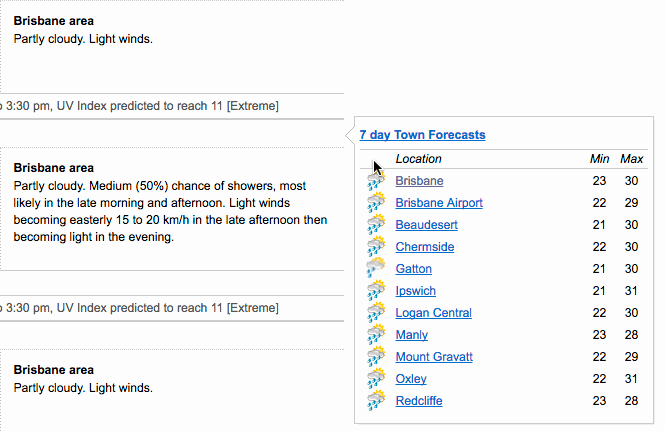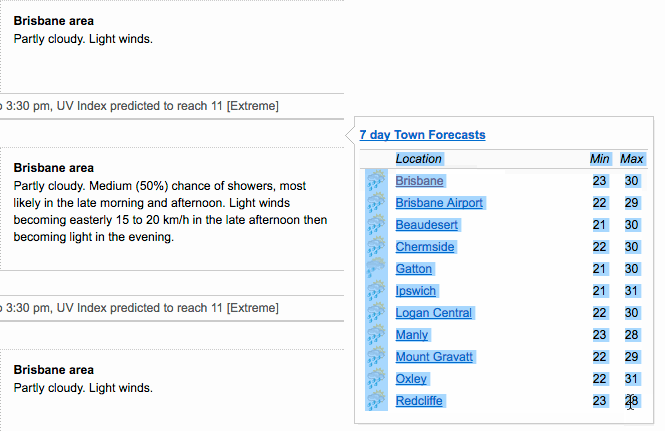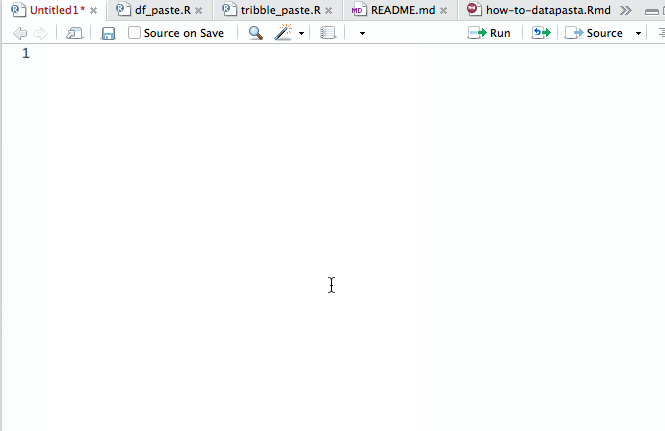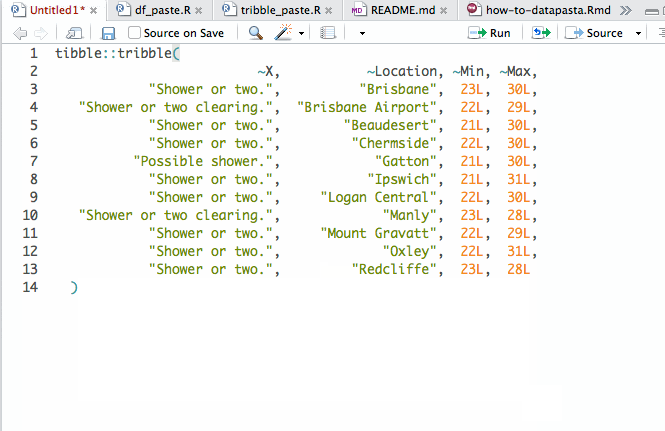
更详细的参考https://cran.r-project.org/web/packages/datapasta/vignettes/how-to-datapasta.html
新年快乐,21年的第一篇文章。
以前写过映射ENSEMBL ID 和 NCBI ID, http://www.zxzyl.com/archives/736。
日常分析中,我们也会经常遇到其他的ID mapping的工作,这种工作不是基因ID转基因ID,而是转录本的ID转基因ID。
如果用的是refGene的注释,最简单了,直接用下面的命令即可
|
|
不过我也经常通过解析gtf文件获得,因为gtf有转本的ID,也有基因的symbol或者ID,只要有gtf文件就可以提取。本着不造轮子的精神,我利用的是现成的R包
|
|
我也在自己的包里面写了一个函数得到ensembl,refseq,hgnc,gene symbol的对应关系,biomaRt比较慢,可以把结果保存成文件
|
|