
Varscan copynumber Recommended Workflow---has beed tested
文章目录
官网提供的推荐流程错误非常多(年代久远??),迄今还没有人详细介绍正确的Varscan copynumber Recommended Workflow。本文改正了官网的recommend workflow,提供正确的pipeline供大家一起学习(我相信我是第一个提供完整流程的哈)。本流程的搭建和解决方案来自网络搜索,感谢万能的网络。本文 没有对命令和输出格式做过多说明,请参阅官方文档。
varscan可以通过配对的肿瘤和组织样本,看覆盖到同一区域内的reads在肿瘤和组织样本中的差异,来检测肿瘤组织中的CNV。
Varscan提供的命令
java -jar varscan.jar copynumber $normal.pileup $tumor.pileup out 或 java -jar varscan.jar copynumber $normal-tumor.mpileup --output-file out
结果如下:
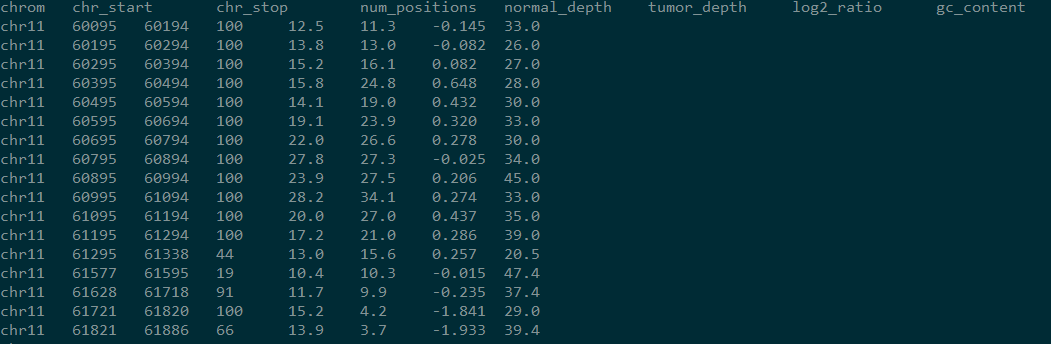
但是该结果只说明了某段区域内,肿瘤和组织depth的差异,区域相连,并没有指明哪个区域是CNV区域,发生了deletion还是insertion等。于是varscan又提供了Recommended Workflow,据说是别人提供给varscan作者的,bug百出惨目忍住。本文会在下文提供正确的流程。在此先提下Recommended Workflow的原理。
原理:
都知道在高通量测序之前,用的是生物芯片。芯片上的每个点都是基因组上的一个marker,通过检测肿瘤样本和组织样本中同一marker荧光强度比值,找到染色体上比值发生改变的位点,然后推荐CNV区域。其中有一种算法,叫做circular binary segment CBS环状二元分割算法(恕我愚笨,不了解这种算法)。R语言包DNAcopy利用CBS和每个marker的lg2ratio,判断那些区域是CNV。那高通量测序varscan的结果如何利用CBS算法呢。从varscan的输出结果可以看出,varscan提供了一定区域内的lg2ratio,Recommended Workflow就将这个区域的起始位点当作这个区域的marker,并与该区域的lg2ratio对应,于是便和芯片检测CNV的方法对接上去了。
流程:
1,Prepare varscan input file
samtools mpileup -f $hgRef $normal > normal.pileup samtools mpileup -f $hgRef $tumor > tumor.pileup
PS:在pileup文件中,如果在某个碱基位置的覆盖度为0,则会缺少对该位点的碱基质量描述的列,导致varscan报错Parsing exception on line。解决该问题,可以在覆盖度为0的碱基那一行添加两个号awk -F"t" ‘{if ($=0)}{print $4"tt*"}‘或者直接过滤该行awk -F"t" ‘$4’。新版本的varscan已经解决处理单样本pileup文件报错的问题,但mpileup文件还是要需要处理的,比如awk -F"t" ‘$4 > 0 && $7 > 0’。
2,Run VarScan copynumber on normal and tumor mpileup output
java -jar varscan.jar copynumber normal.pileup tumor.pileup out
3,Run VarScan copyCaller to adjust for GC content and make preliminary calls
java -jar varscan.jar copyCaller out.copynumber --output-file out.copynumber.called
PS:芯片时需要adjust,因为需要校正系统误差,同理,此步也是应该的。到此步位置,步骤都还正常。
4,Apply circular binary segmentation using the DNAcopy library from BioConductor
source("http://bioconductor.org/biocLite.R")
biocLite("DNAcopy")
library("DNAcopy")
## header 一定要设为T,因为有header。
cn <- read.table("out.copynumber.called",header=T)
pos <- round((cn[,2]+cn[,3])/2)
CNA.object <-CNA( genomdat = cn$adjusted_log_ratio, chrom = cn[,1], maploc = pos, data.type = 'logratio', sampleid=c("$sampleName"), presorted=TRUE)
CNA.smoothed <- smooth.CNA(CNA.object)
segs <- segment(CNA.smoothed, verbose=0, min.width=2)
out=segments.p(segs)
write.table(out, file="out.copynumber.called.seg", row.names=F, col.names=T, quote=F, sep="t")
perl mergeSegments.pl out.copynumber.called.seg --ref-arm-sizes armsize.txt --output out
结果如图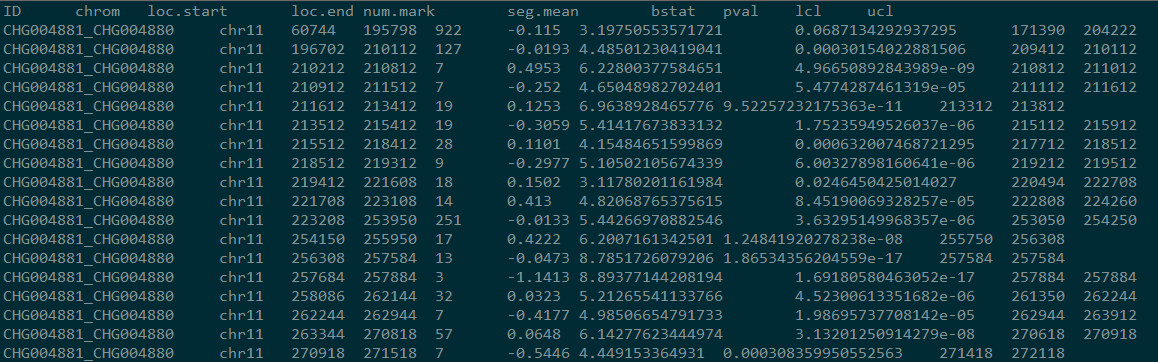
5,Merge adjacent segments of similar copy number and classify events by size (large-scale or focal)
这一步也很坑人,mergeSegments.pl提到
–ref-arm-sizes Two column file of reference name and size in bp for calling by chromosome arm
看清楚,是两列,然而正确的格式应该是四列!!如下:
chr1 0 125000000 p chr1 125000000 249250621 q
然而,还没有完,运行接着报错Use of uninitialized value $stats{“num_merged_events”} in array element at mergeSegments.pl line 182, line 3. 好吧,要在mergeSegments.pl文件中第109行初始化num_merged_events变量。还没有完,报错说$sample没有,DNAcopy的结果中确实没有啊,那只好把134行这个变量删掉了。然后才能正确运行,运行命令如下:
perl mergeSegments.pl out.copynumber.called.seg --ref-arm-sizes armsize.txt --output out
会生成两个文件,一个summary,一个events,events文件截图如下: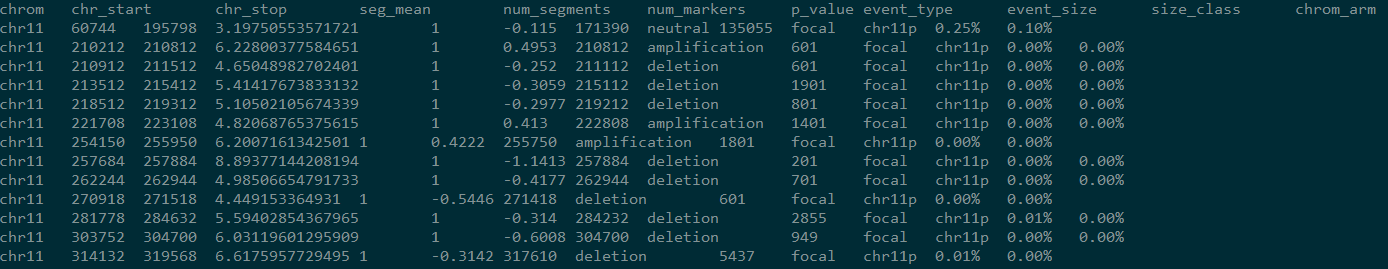
前三列表示 CNV 发生的区域
第六列为校正后的 log2 fold change 值,正值表示扩增,负值代表减少
第八列为 CNV 发生的状态,为 deletion,neutral 或者 amplification
附件:
arm size文件:arm size
修改后的mergeSegments.pl文件:mergeSegments.pl
附流程:
|
|
Ref:
http://dkoboldt.github.io/varscan/copy-number-calling.html#copy-number-workflow
BioStar
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2016-01-26