分析主要基于Rstudio Server的Rmarkdown,这样在线版的Rstudio可以无缝和服务器协同,Rmarkdown管理代码也方便。但免费版的Rstudio Server也有痛点:(1)不容易切换R,必须管理员统一设置,要变所有用户都变;(2)不能多开,只能打开一个Rstudio Server的页面。当然痛点也能解决,就是用Pro版本,最少要4,975(美刀)。
后来Rstudio的公司改名为Positron,又出了Positron IDE,运行R和python比vscode 方便,比如图片展示,变量查看,接近Rstudio,但不支持remote ssh,意味着我不能用远程服务器上的R。所以尝试了下就不在使用。
再后来Positron支持了remote ssh,但是AI这块的支持还是不如vscode,体验了一把又不用了。
再再后来,Positron支持了AI assistant,我又开始尝试用Positron。基于这次体验,我决定后续把Positron当主力了。
- 对Rmardown的支持:我严重依赖Rmarkdown,特别是项目的函数多的时候,我分代码块,便于查找内容和管理。Postrion默认就支持R和Rmarkdown。特变是Rmarkdown的Table of Contents,可以快速跳转到不同代码块,Positron本来就基于vscode的壳,自带了outline的内容框,可以显示markdown的标题。
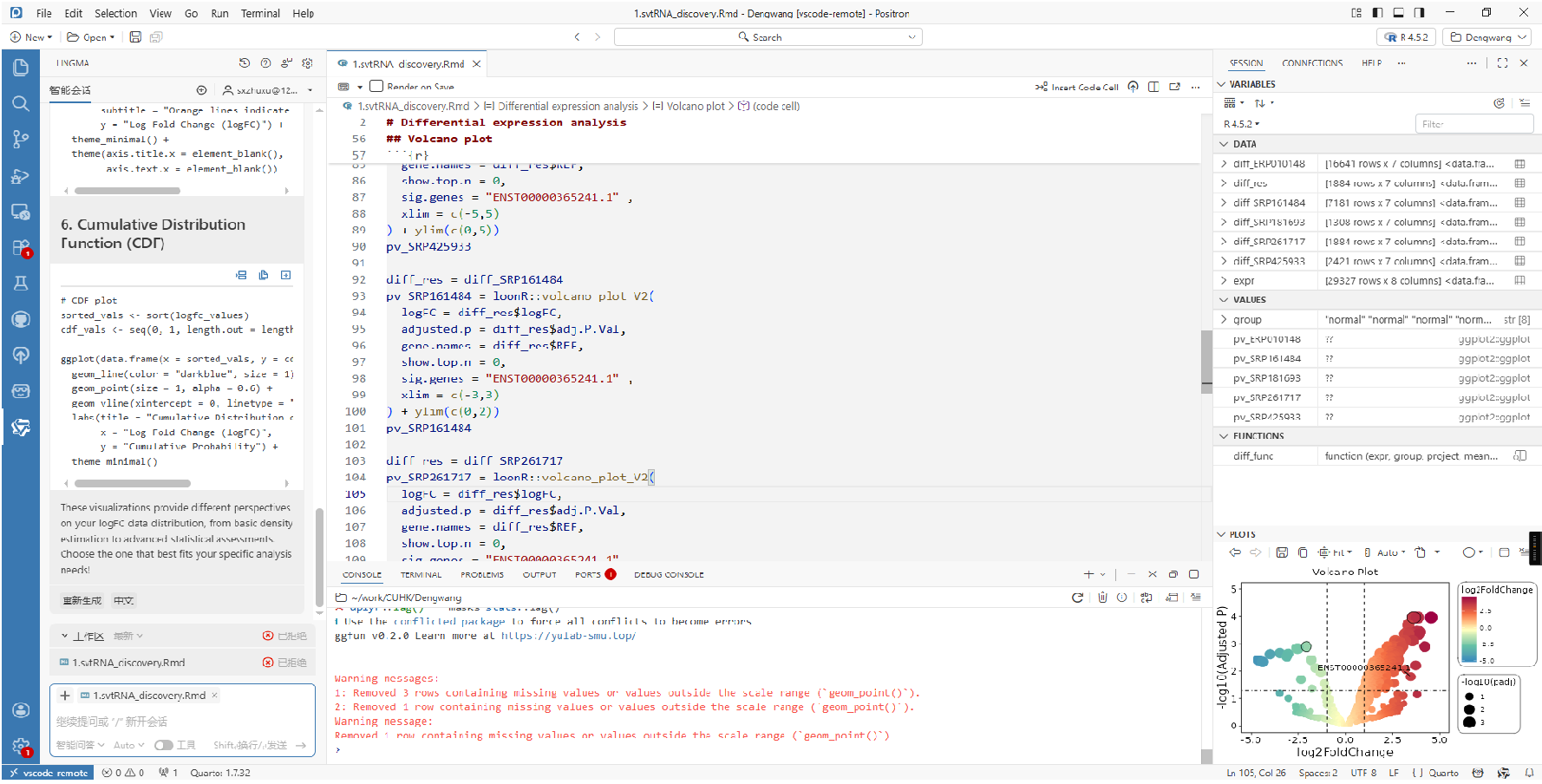
- 变量和图片:我不喜欢vscode的原因就是变量不方便查看,图片不想Rstudio那样专门有个panel展示,Postiron的变量查看和图片展示逻辑和Rstudio一样,有专门的panel。
- 自动补全:我下载了千问的灵码和字节的Marscode,也试了自带的copilot,尝试行内自动补全的功能,感觉真的太好了,可以通过上下文,把我想写的代码都提示出来,不仅仅是传统IDE那种函数提示,效率大大提高。Marcode支持jupyter,但不支持Rmarkdown,灵码支持Rmarkdown,不支持jupyter。如果支持quarto的话,我一定会切到quarto格式。
感觉不如Rstudio的地方:(1)图片的展示效果,不知道为什么感觉没有Rstudio好看;(2)数据框里面的数据搜索,我以前直接在搜索框搜索即可,Positron需要选定特定列,添加filter,稍微麻烦点;(3)有时候还是会报错,比如LSP服务断开,或者画图失败。
关于Rmarkdown:我个人认为比jupyter方便,因为我可以在代码块运行,也可以把代码放到同一环境下的console里面。Rmarkdown绝对是我在Rstudio/Positron生态里的重要因素,现在的qmd和rmd都是类似,Positron主推qmd。可惜的是两年前Rmarkdown贡献者xieyihui被Positron解雇了。如果灵码或者Marscode支持qmd的话,我可能会用qmd写代码。