
What is a Scaffold?
A scaffold is a portion of the genome sequence reconstructed from end-sequenced whole-genome shotgun clones. Scaffolds are composed of contigs and gaps. A contig is a contiguous length of genomic sequence in which the order of bases is known to a high confidence level. Gaps occur where reads from the two sequenced ends of at least one fragment overlap with other reads in two different contigs (as long as the arrangement is otherwise consistent with the contigs being adjacent). Since the lengths of the fragments are roughly known, the number of bases between contigs can be estimated.
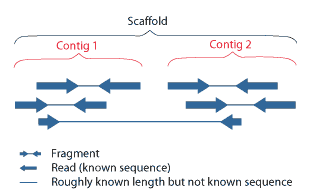
The goal of whole-genome shotgun assembly is to represent each genomic sequence in one scaffold; however, this is not always possible. One chromosome may be represented by many scaffolds (e.g., Chlamydomonas reinhardtii) or just a single scaffold (e.g., Human chromosome 19), depending on how completely the genome can be reconstructed, or assembled, from the available reads. The relative locations of scaffolds in the genome are unknown.
N50 explained
In computational biology, the N50 statistic is a statistic of a set of contig lengths. The N50 is similar to a mean or median, but has greater weight given to the longer contigs. It is used widely in genome assembly, especially in reference to contig lengths within a draft assembly. Given a set of contigs, each with its own length, the N50 length is defined as the length for which the collection of all contigs of that length or longer contains at least half of the total of the lengths of the contigs, and for which the collection of all contigs of that length or shorter contains at least half of the total of the lengths of the contigs. (When more than one value of length meets both these criteria then the N50 is the average of the longest and shortest lengths that meet these criteria.) This can be thought of as the point of half of the mass of the distribution; the number of bases from all contigs shorter than the N50 will be close to equal to the number of bases from all contigs longer than the N50. The N90 statistic is smaller than or equal to the N50 statistic; it is the length for which the collection of all contigs of that length or longer contains at least 90% of the total of the lengths of the contigs, and for which the collection of all contigs of that length or shorter contains at least 10% of the total of the lengths of the contigs.
Getting LibSVM to work with Weka on Mac(reprint)
Somehow, the only way to use LibSVM with Weka is by using the bash command-line. I have tried the second method successfully. As for you , etheir is good.
Step 1: Get Weka.Assume the bleeding edge version 3.7.0. Unzip and put in /Applications folder.
Step 2: Get LibSVM.
a. Iowa State site (http://www.cs.iastate.edu/~yasser/wlsvm/wlsvm.zip):
If you use Safari to download, it will be unzipped in the Downloads directory. The files you need are ~/Downloads/WLSVM/lib/wlsvm.
How to extract paired-end reads from SRA files(reprint)
SRA(NCBI) stores all the sequencing run as single “sra” or “lite.sra” file. You may want separate files if you want to use the data from paired-end sequencing. When I run SRA toolkit’s “fastq-dump” utility on paired-end sequencing SRA files, sometimes I get only one files where all the mate-pairs are stored in one file rather than two or three files. The solution for the problem is to always run fastq-dump