解决 mount: unknown filesystem type ntfs
移动硬盘是ntfs格式的,服务器不能mount,报错 mount: unknown filesystem type ‘ntfs’
解决方法:安装 NTFS-3G,官网 https://www.tuxera.com/community/open-source-ntfs-3g
安装
|
|
挂载
|
|

移动硬盘是ntfs格式的,服务器不能mount,报错 mount: unknown filesystem type ‘ntfs’
解决方法:安装 NTFS-3G,官网 https://www.tuxera.com/community/open-source-ntfs-3g
安装
|
|
挂载
|
|
Should I switch to a newer reference?
GRCh38 consists of several components: chromosomal assembly, unlocalized contigs (chromosome known but location unknown), unplaced contigs (chromosome unknown) and ALT contigs (long clustered variations). The combination of the first three components is called the primary assembly. It is recommended to use the complete primary assembly for all analyses.
参考基因组包括chromosomal assembly, unlocalized contigs (chromosome known but location unknown), unplaced contigs (chromosome unknown)和ALT contigs (long clustered variations),前三个是primary assembly,alt contigs代表的是部分区域单体型的多样性,这些区域过于复杂不能用一条序列表示。
In addition to adding many alternate contigs, GRCh38 corrects thousands of SNPs and indels in the GRCh37 assembly that are absent in the population and are likely sequencing artifacts. It also includes synthetic centromeric sequence and updates non-nuclear genomic sequence.
除了添加了很多alternative contigs,GRCh38更正了数以千计的GRCh37版本中的SNPs和indels,这些SNPs和indels在人群中没有出现过、可能因为测序错误导致。GRCh38版本也包含了人工的着丝粒序列和更新了非核基因组序列。
The ability to recognize alternate haplotypes for loci is a drastic improvement that GRCh38 makes possible. Going forward, expanding genomics data will help identify variants for alternate haplotypes, improve existing and add additional alternate haplotypes and give us a better accounting of alternate haplotypes within populations. We are already seeing improvements and additions in the patch releases to reference genomes, e.g. the seven minor releases of GRCh38 available at the time of this writing. GRCh38大幅提高了识别alternate haplotype的能力,进一步提高了识别alternate haplotype的突变的能力。
BWA的作者Li Heng推荐GRCh37 primary assembly+ALT+decoy组成的参考基因组hs38DH,可以通过bwa下载,见https://github.com/lh3/bwa/blob/master/README-alt.md :
bwa.kit/run-gen-ref hs38DH
作者的用NA12878做测试的比对结果如下,
|
|
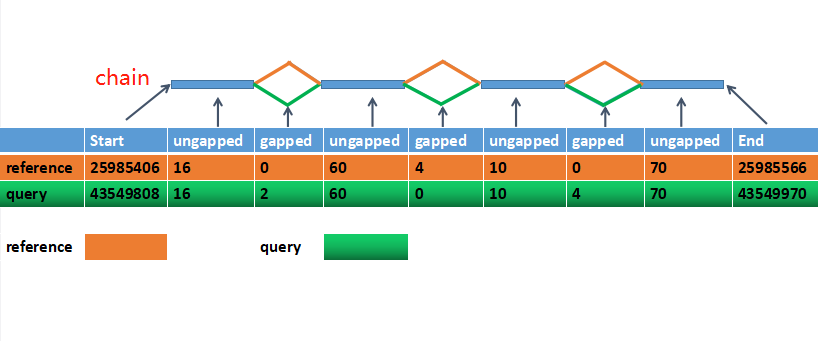
在做基因组坐标转换的时候,用crossmap和liftover的时候,会用到chain file。大家都在讲坐标转换会用到这个文件,却没有讲过chain文件的具体内容(百度和谷歌搜索结果都没有中文介绍,本文应该是第一个)。本文的内容都翻译自UCSC网站,原文https://genome.ucsc.edu/goldenpath/help/chain.html,希望能帮到大家了解这个文件。
UCSC chain文件 http://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/ Ensembl chain文件 https://sourceforge.net/projects/crossmap/files/Ensembl_chain_files/
chain file里面包含许多块alignment的信息(个人觉得可以理解为同源的地方,chain),其中每一块有一个header,记录alignment在两个版本中坐标,以及许多行alignment data line记录具体比对情况。
即每一块有 Header Line 和 Alignment Data Lines组成。形式如下,有两个chain
|
|
分析时使用的基因组版本,可能会与其他来源数据所使用的基因组版本不一致,需要统一成同一个版本的坐标,才能方便下一步的分析。
常用的有NCBI的Remap在线服务和UCSC的liftover,其实还有很多,本文暂时总结部分工具的用法。以将APOA1的编码区坐标(利用UCSC的genome browser下载,或者下载该文件APOA1.bed)转换为例,从hg19转到hg38版本坐标上。需要注意的是,在使用的时候,需要注意是否支持对应的格式。
| 类型 | 支持格式 | 地址 | 推荐指数 | |
|---|---|---|---|---|
| Liftover | 在线 | bed | http://genome.ucsc.edu/cgi-bin/hgLiftOver | 一般 |
| Liftover | 本地 | bed和gff | http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/liftOver | 推荐 |
| Remap | 在线 | hgvs,bed,gvf,gff,gtf,Text ASN.1,Binary ASN.1,UCSC Region和VCF | https://www.ncbi.nlm.nih.gov/genome/tools/remap | 推荐 |
| CrossMap | 本地 | SAM/BAM,,Wiggle/BigWig, bed, gff/gtf,VCF | http://crossmap.sourceforge.net/ | 推荐 |
| picard | 本地 | interval和VCF | http://broadinstitute.github.io/picard/ |
大家经常用UCSC的hg19和NCBI的GRCh37版本的,但还有其他的版本,比如b37,hg37d5,比如在分析NIST的genome in a bottle(GIAB)提供的bam数据时,就遇到了hg37d5的版本,在用GATK的时候会遇到b37版本。
Genome Reference Consortium(基因组参照序列联盟),由英国Wellcome Trust Sanger研究中心(the Wellcome Trust Sanger Center)、华盛顿大学基因组中心(The Washington University Genome Center)、欧洲生物信息研究所(the European Bioinformatics Institute)和美国国家生物技术信息中心(NCBI)联合组成。
GRCH37版本发布之后,也会有小的更新,比如GRCh37.p2,大的更新比如由GRCh37升级到GRCh38,填补gap,修改部分序列,其目的是提供一个完整的基因组序列assemble。GRCh38已经在2013年发布,多数基因组数据库正在兼容或者更新到该版本。
该版本包含人类chr1到chr22,chrX,chrY,MT染色体以及
下载地址:ftp://ftp.ncbi.nih.gov/genomes/Homo_sapiens