BCFTOOLS支持的表达式
翻译https://samtools.github.io/bcftools/bcftools-man.html#expressions 有效的

此时VCF文件中的突变,与刚开始下机得到的FASTQ文件类似,称为raw data。此时的突变集合中,有很多假阳性突变,这些突变需要在突变分析之前过滤掉。
传统的过滤方式,直接根据每个突变的注释信息,进行过滤。最直接和最常见的是根据DP标签过滤,即根据该突变位点的测序深度进行过滤。通常,深度越低,支持该突变的reads数目越少,该突变越不可信。还可以根据前面提到的QUAL质量分值进行过滤,分值越低越不可信。Forward reads和Reverse reads的比例。通过,设定一定的阈值,看这些注释信息是高于还是低于该阈值。
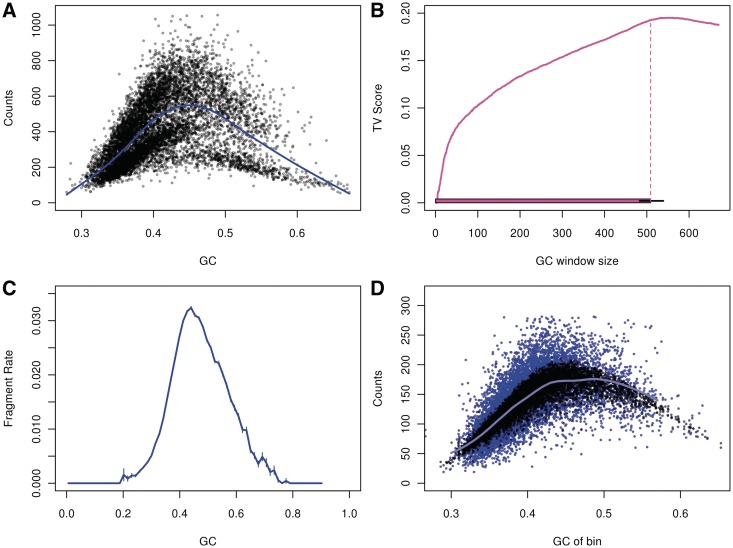
测序中的GC偏好指的是基因组上GC含量在50%左右的区域更容易被测到,产生的reads更多,这些区域的覆盖度更高,在高GC或者低GC区域,不容易被测到,产生较少的reads,这些区域的覆盖度更少。用基因组单位长度的bin中的GC含量作为横坐标,覆盖度作为纵坐标作图,可以明显的看到该趋势。这种趋势在100kb为单位的bin中依然存在。
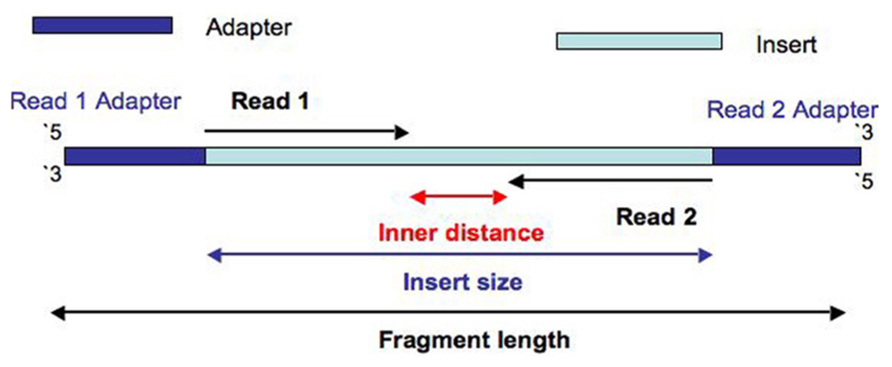
**Insert_size = the sequence betwwn adapters **
Fragment_size= Insert_size + Adapters
测序得到的原始测序序列,里面含有低质量的reads。低质量的reads可能因为flowcell上的cluters不是有单一DNA扩增而来,或者几个cluters混为一起等。还有测序仪在前几个和几个cycle测序质量不好,需要关注一条read的前后几个碱基的质量。如果质量非常不好,测出来的碱基可能为N(无法确定碱基类型)。
此外,原始reads中还包含测序接头等序列。如果一个文库的平常插入长度为450bp的话,不一定每个插入长度都为450bp,如果个别分子插入长度为100bp,双端配对150bp测序的,会将该片断测穿,配对的reads会多包含50bp的index或者SP等序列。为了保证信息分析质量,需要对下机的raw reads 进行精细过滤,得到clean reads,后续分析都基于clean reads进行。
为了提高下一步的比对质量,此时数据预处理的过程主要包括: • 去掉接头,去掉开头和结尾几个碱基中质量不好的碱基 • 滑窗扫描,检查是否有好几个连续碱基质量不好的情况 • 丢弃过短的read • 去接头 • 去掉前端碱基质量低于一定值的碱基 • 去掉后端碱基质量低于一定值的碱基 • 以4bp为窗口滑窗扫描read,如果4个碱基平均质量低于15,则截断 • 丢弃序列长度小于36bp的reads