
CoNVaDING和DECoN简评--基于Panel测序的外显子拷贝数变异分析
文章目录
目的
外显子水平的拷贝数变异和许多疾病有关系,需要检测外显子水平拷贝数变异。
需要解决的实践问题
1)成本问题:如果利用原有测序数据(全外显子测序数据或panel 测序),而不重复进行实验,实现一次测序,解决多种问题 2)灵敏度和特异性的问题:希望在高灵敏度的情况下,获得尽可能高的特异性 3)分辨率:需要外显子水平而非基因组范围内的拷贝数变异
样本
我们测了Coriell的已知有特定基因拷贝数变异的样本作为阳性样本,把正常人的样本作为对照样本
工具
随着分析技术的发展,针对外显子水平的分析工具开始出现,CoNVaDING、DECoN、PureCN、panelcn.MOPS、ExomeDepth、CODEX2,这些软件都利用了外显子区域的覆盖度信息,用参考基因组GC含量校正,然后根据不同算法来识别拷贝数变异。 从中选择了两种最新的软件CoNVaDING和DECoN。因为这两种软件不需要配对样本数据,只需提供实验样本和对照样本组即可,另外这两个软件较新。 CoNVaDING利用一组可能的对照样本,并从中选择模式pattern最相似的样本作为对照样本,并对每个基因所有目标区域的depth进行标准化,通过比较阳性样本和对照样本之间计算z score和ratio score判断外显子是否发生拷贝数变异。DECoN则是对ExomeDepth工具进行了优化,而ExomeDepth利用贝塔二项分布来描述特定区域正常样本和对照样本的覆盖度比值,用隐马尔可夫模型来预测。
DECoN 1.0.1 外显子拷贝数检测软件 https://github.com/RahmanTeam/DECoN CoNVaDING 1.2.0 外显子拷贝数检测软件 https://github.com/molgenis/CoNVaDING
结果
我们做了很多工作来验证拷贝数分析工具的性能,涉及数据问题,只能简略的讨论一下CoNVaDING和DECoN的性能。
<灵敏度>
灵敏度 PPA = Sensitivity = True positive/(True positive + false negative)
我们的多个拷贝数阳性样本,总共有22个外显子DUP和40个外显子DEL(分布在不同阳性样本上,且区域不尽相同)。各个流程的结果如下图,
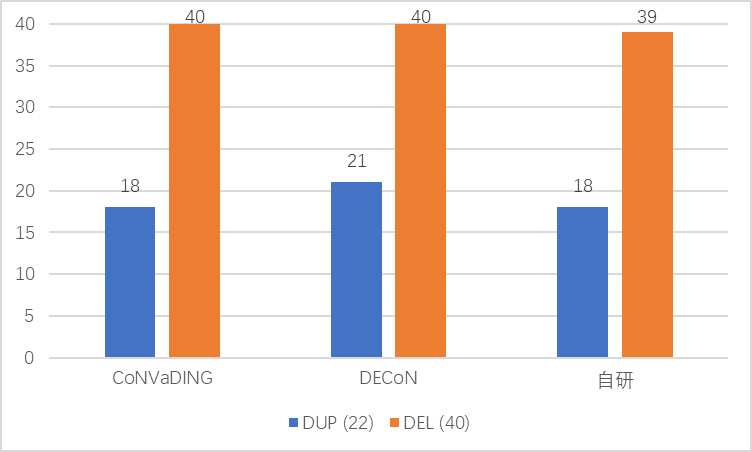
可以看出CoNVaDING、DECoN和自研流程都将大部分外显子水平的拷贝数变异检测出,特别是DEL变异。检测性能最好的是DECoN,40个DEL变异全部检测出,而22个DUP变异中检测出21个。我们自己研发的流程和CoNVaDING都检测出了22个DUP变异中的18个。这说明检测的灵敏度尚可,且DECoN最佳。
<特异性>
如果我能检测出拷贝数变异,但是存在于100个假阳性中,从这么多假阳性变异中找到真的拷贝数变异无疑是一项巨大的工作。所以我们要尽可能的提高特异性。
特异度 PPV = Specificity = True negative/(True negative + false positive)
各流程的灵敏度和特异性结果如下图
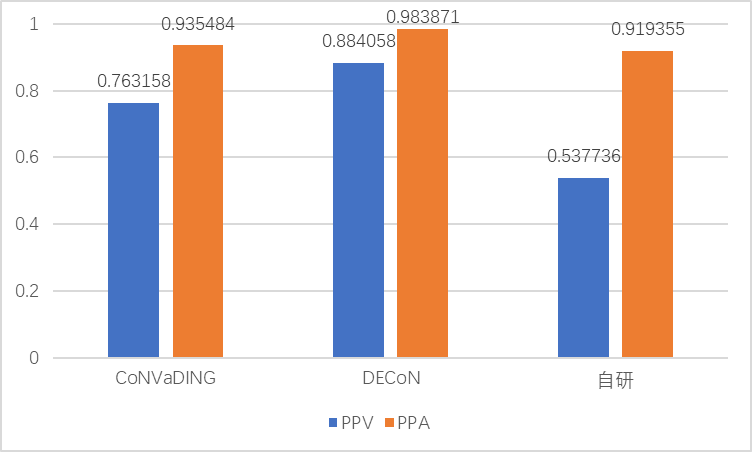 可以看出,DECoN的性能又是最佳,特异性达到了88%。自研流程因为缺陷问题,PPV最小。
可以看出,DECoN的性能又是最佳,特异性达到了88%。自研流程因为缺陷问题,PPV最小。
<其他>
我们还比较了对照样本数目对外显子拷贝数变异分析的影响,比较了DUP和DEL类别下的各流程的灵敏度和特异性。结果均显示DECoN可以在少量对照样本的情况下,达到分析要求,展示出最优异的性能。
结论和讨论
DECoN与CoNVaDING比起来,DECoN的性能更加,可以用来检测外显子水平的拷贝数变异。 1)在分析外显子拷贝数变异时应增加对照样本数目,提高PPV和PPA指标。此外在挑选对照样本时,应尽量选择与阳性样本同一测序平台、同一测序批次、相近数据量的样本。 2)在分析特定基因的外显子拷贝数变异时,不应只提供目标基因的区域,而应提供更多的额外基因区域,供软件来分析基因覆盖度的范式pattern,以提高软件分析目标区域外显子拷贝时的性能。
背景
拷贝数变异是结构变异的一种,研究已经证实拷贝数变异与人类疾病的发生相关,例如智力缺陷、自闭症、精神分裂、癌症发生等。不同于基因的单碱基变异,外显子水平的拷贝数变异(Exon Copy Number Variant)是一类不常见但非常重要的突变类型,约10%的BRCA1癌症是由外显子拷贝数变异引起的。典型的外显子拷贝数变异可能导致蛋白紊乱甚至丧失功能。过往检测拷贝数变异的方法是利用多重连接探针扩增技术(multiplex ligation-dependent probe amplification ,MLPA),染色体芯片(CMA)或荧光PCR。随着测序技术的发展,利用NGS的数据分析基因拷贝数变化是越来越受到关注和有效的确定基因拷贝数的方法。利用同一套数据,在检测单碱基突变和插入缺失的同时,检测外显子拷贝数的变异,是一快速、低成本的方式。 通过目标区域测序进行外显子拷贝数分析有如下挑战:
1)常规通过测序得到拷贝数变异,往往是基于全基因组测序,因为全基因组测序是DNA随机打断建库,覆盖度较为均一,而外显子测序或panel测序存在捕获效率等问题,覆盖度均一性较全基因组差;
2)基于全基因组测序的拷贝数变异分析检测一般侧重于大片段序列的缺失或重复(通常大于1Mbp),而外显子拷贝数变异的序列长度在100bp左右;
3)多数软件是基于全外或全基因组测序,而非panel测序。
流程
CoNVaDING分析流程:
- 统计覆盖度Create normalized count files
- 选择代表性对照样本Selecting the most informative control samples
- 外显子拷贝数变异检测CNV Detection
DECoN分析流程:
- 统计覆盖度Reading BAM files to generate coverage metrics
- 外显子拷贝数变异检测Calling exon CNVs
|
|
如果你的数据染色体编号是1、2、3而不是chr1、chr2、chr3,可以开启–custom TRUE并提供–exons customNumbers.file transProb是隐马尔可夫模型的状态转换概率,可以自己设置,该值越大,拷贝数变异检测的灵敏度越高。
自研分析流程:
- 统计覆盖度
- 外显子覆盖度标准化
- 外显子拷贝数变异预测
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2018-04-25