
bismark DNA甲基化测序比对-bisulfite-seq
文章目录
bismark调用bowtie2进行比对,调用samtools生成bam文件,因此在运行bismark之前,需要安装bowtie2和samtools
请注意,fastq文件要进行质控,比如去掉低质量的reads,去掉adaptor等,可以看本文最下方推荐的PPT,本文不介绍,此外本本只介绍到序列比对,后续的统计分析没有介绍,有兴趣的朋友可以关注swDMR和methykit工具包。
安装bismark
|
|
生成转换后的基因组
# --path\_to\_bowtie后面跟的是文件夹
# --verbose 输出log信息
# ./ref 文件夹中有一个基因组fasta文件
# --bowtie2指明用的是bowtie2
./bismark\_v0.16.1/bismark\_genome\_preparation --path\_to_bowtie /home/zzx/bowtie2-2.2.9/ --bowtie2 --verbose ./ref/
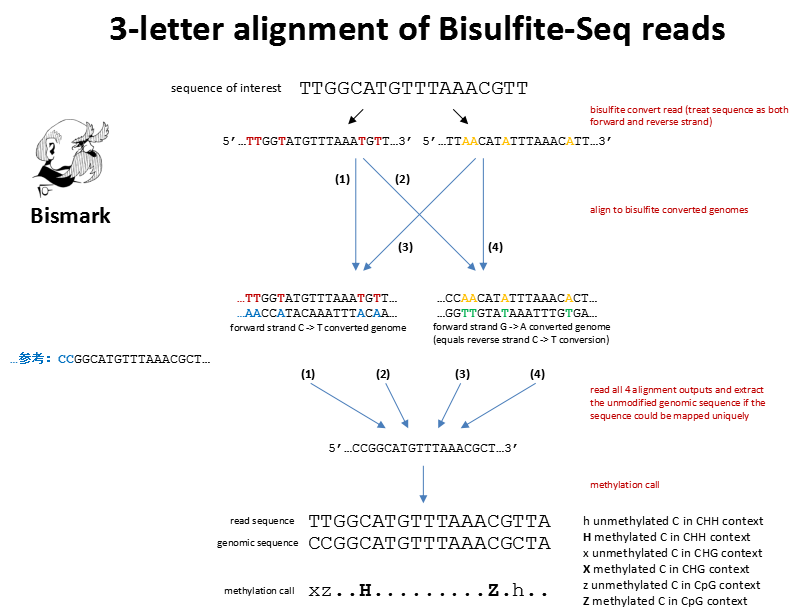
因为在重亚硫酸盐甲基化测序中,因为未甲基化的C会变为T,在正链表现为C–>T,但是在负链有C变为T,转换为正链时,即为G–>A,所以基因组需要进行两种转化,才能用于比对。在基因组目录下产生Bisulfite_Genome目录,有CT_conversion和GA_conversion文件夹,这两个文件夹包含转换后的fasta文件和bowtie2建立的索引bt2文件。
fastq中的BS转换后的read与转换的参考基因组比对,得到在参考基因组中的位置,再与原始的参考基因组比较,确定methylate call
bismark比对
--non_bs_mm Optionally outputs an extra column specifying the number of non-bisulfite mismatches a read during the alignment step.
--nucleotide_coverage 该选项 会生成nucleotide_stats.txt文件。creates a '...nucleotide_stats.txt' file that is also automatically detected by bismark2report and incorporated into the HTML report.
--multicore Sets the number of parallel instances of Bismark to be run concurrently. 一个实例已经将1个核分配给bismark,两个或者4个核分配给bowtie2,一个分给samtools
./bismark\_v0.16.1/bismark --genome\_folder ./ref --bowtie2 --nucleotide\_coverage --non\_bs\_mm --basename test --temp\_dir ./tmp --samtools\_path ./samtools-1.2/ --path\_to\_bowtie ./bowtie2-2.2.9/ -1 1.fastq -2 2.fastq --output\_dir ./
生成bam文件,bam文件可以用samtools view转成sam格式
SAM文件
ST-E00206:135:HMMMWCCXX:4:1101:3710:1801\_1:N:0:GATCAG 163 chr13 20584212 40 90M = 20584274 183 TTTTCCATATTTTAAACTTTTAATTTTTCTTATAAATATCTTAATACATATACGATAATATCTCATAATTTTAATTTACATTTCCCTAAT A,<FFKAKKKKKKA,AFF<FK,<AFKKKAKK<AAF<KA<<FKF<F,,7FFFA,<<AA<K,<AFA7AA,FKKKA7FKF7A7AKKK,AA,A7 NM:i:16 MD:Z:8G6G5G9G2G8G1G3G1G2G1G0G8G0G4G4G12 M:Z:........h......h.....h.........h..h........h.h...h.h.Zx.hh........hh....h....h............ XR:Z:GA XG:Z:GA XB:Z:0
(11) QUAL (Phred33 scale)
(12) NM-tag (edit distance to the reference)
Edit distance to the reference, including ambiguous bases but excluding clipping NM-tag,与bowtie中NM:i相同。read string转换成reference string需要的最少核苷酸的edits:插入/缺失/替换
(13) MD-tag (base-by-base mismatches to the reference)
String for mismatching positions
eg.XX:Z:2C4C2C6C1C1AC18C3C15C4C2CC14
表示2个碱基完全匹配,一个C替换,接着4个碱基完全匹配,一个C发生替换.
(14) XM-tag (methylation call string)
methylation call:
X for methylated C in CHG context (was protected) CHG上的C发生了甲基化
x for not methylated C CHG (was converted) CHG上的C未发生甲基化
H for methylated C in CHH context (was protected) CHH上的C发生了甲基化
h for not methylated C in CHH context (was converted) CHH上的C未发生甲基化
Z for methylated C in CpG context (was protected) CpG上的C发生了甲基化
z for not methylated C in CpG context (was converted) CpG上的C未发生甲基化
. for any bases not involving cytosines
(15) XR-tag (read conversion state for the alignment)
共两种转换:CT和GA,GA就是指将read里的所有G转换成A
eg.XR:Z:CT
(16) XG-tag (genome conversion state for the alignment)
共两种:GA和CT。
CT是指将全基因组上所有的C转换成T
eg.XG:Z:CT
此外还会生成一个比对的PE_report.txt文件,后续生成html版本的report会用到
deduplicate 去重
会生成deduplicated.bam为后缀的文件
|
|
提取甲基化信息
bismark_methylation_extractor --no_overlap --paired-end --bedGraph --comprehensive --counts --remove_spaces --buffer_size 10G --cytosine_report --genome_folder ref/ --output ./test_pe.deduplicated.bam
–remove_spaces Replaces whitespaces in the sequence ID field with underscores to allow sorting.
–cytosine_report 指报道全基因组所有的CpG。只有当指定
–cytosine_report时才需要genome_folder。生成的文件很大 After the conversion to bedGraph has completed, the option
–cytosine_report produces a genome-wide methylation report for all cytosines in the genome. By default, the output uses 1-based chromosome coordinates (zero-based cords are optional) and reports CpG context only (all cytosine context is optional). –bedGraph 指将产生一个BedGraph文件存储CpG的甲基化信息
–counts 指在bedGraph中有每个C上甲基化reads和非甲基化reads的数目 -
-genome_folder 后跟着参考基因组的位置
–no_overlap Suppresses the Bismark version header line in all output files for more convenient batch processing. This option avoids scoring overlapping methylation calls twice (only methylation calls of read 1 are used for in the process since read 1 has historically higher quality basecalls than read 2).
–paired-end Input file(s) are Bismark result file(s) generated from paired-end read data. –comprehensive 指把四条链的结果合并为一个文件 Specifying this option will merge all four possible strand-specific methylation info into context-dependent output files.
–cutoff [threshold] The minimum number of times a methylation state has to be seen for that nucleotide before its methylation percentage is reported. Default: 1 (i.e. all covered cytosines) bismark extractor不支持sort后的bam文件,可能因为sort之后,配对的read不在连续的两行。 This might be a result of sorting the paired-end SAM/BAM files by chromosomal position which is not compatible with correct methylation extraction. Please use an unsorted file instead
–no_header Suppresses the Bismark version header line in all output files for more convenient batch processing
–CX/-CX_context bedGrap中包含CpG context以外的
CHG_context_test_pe.txt CHH_context_test_pe.txt
CpG_context_test_pe.txt
CpG_context_test_pe.txt.spaces_removed.txt
这四个context文件格式如下
(1) seq-ID
(2) methylation state + 为甲基化, - 为未甲基化
(3) chromosome
(4) start position (= end position)
(5) methylation call
test_pe_splitting_report.txt
test_pe.M-bias.txt R1和R2中M-bias作图的数据
test_pe.M-bias_R1.png M-bias图
test_pe.M-bias_R2.png M-bias图
test_pe.bedGraph.gz 0-based genomic start and 1-based end coordinates
test_pe.bismark.cov.gz
By default, this mode will only consider cytosines in CpG context, but it can be extended to cytosines in any sequence context by using the option ‘–CX’
生成report
|
|
会生成一个html文件,可以在浏览器中打开
参考:
http://blog.sciencenet.cn/home.php/fgcfmt/home.php?mod=space&uid=1271266&do=blog&id=842711
www.bioinformatics.babraham.ac.uk/projects/bismark/
http://www.bioinformatics.babraham.ac.uk/training/Methylation_Course/BS-Seq%20theory%20and%20QC%20lecture.pptx 强烈推荐阅读该PPT,介绍了甲基化测序,质控,比对,去重等
http://www.bioinformatics.babraham.ac.uk/projects/bismark/Bismark_User_Guide.pdf
##################################################################### #版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任 #Author: Jason
###################################################################
文章作者 zzx
上次更新 2016-05-21