
JC-单细胞转录组分析揭示人类子宫内膜癌的起源和病理过程
文章目录
Single-cell transcriptomic analysis highlights origin and pathological process of human endometrioid endometrial carcinoma
https://www.nature.com/articles/s41467-022-33982-7
背景
子宫内膜癌(Endometrial cancer, EC)是妇科最常见的恶性肿瘤之一,子宫内膜样子宫内膜癌(endometrioid endomecancer, EEC)是EC的主要病理类型。
在雌激素依赖性EEC肿瘤发生过程中,子宫内膜在没有孕激素保护的情况下长期暴露于雌激素中,表现出不受控制的增殖,并且可以从正常子宫内膜发展到非典型子宫内膜增生(AEH, EEC癌前阶段),然后逐步发展到EEC。关于ECC的起源过往研究推测包括子宫内膜上皮和基质干成分在内的多种谱系可能是EEC的起源,但证据不足以支持明确起源。
肿瘤微环境由免疫细胞、成纤维细胞、周细胞等组成,在肿瘤的发生、预后和转移中起重要作用,尽管先前的研究已经提示肿瘤微环境在预后和治疗耐药的潜在作用,但从正常子宫内膜到EEC形成的过程仍不明确。
|
|
结果
正常子宫内膜、AEH和EEC的细胞图谱
- 划分了6个亚群(上皮细胞,间质成纤维细胞、内皮细胞、淋巴细胞、巨噬细胞、平滑肌细胞),其中上皮细胞又细分为纤毛上皮和非纤毛上皮。
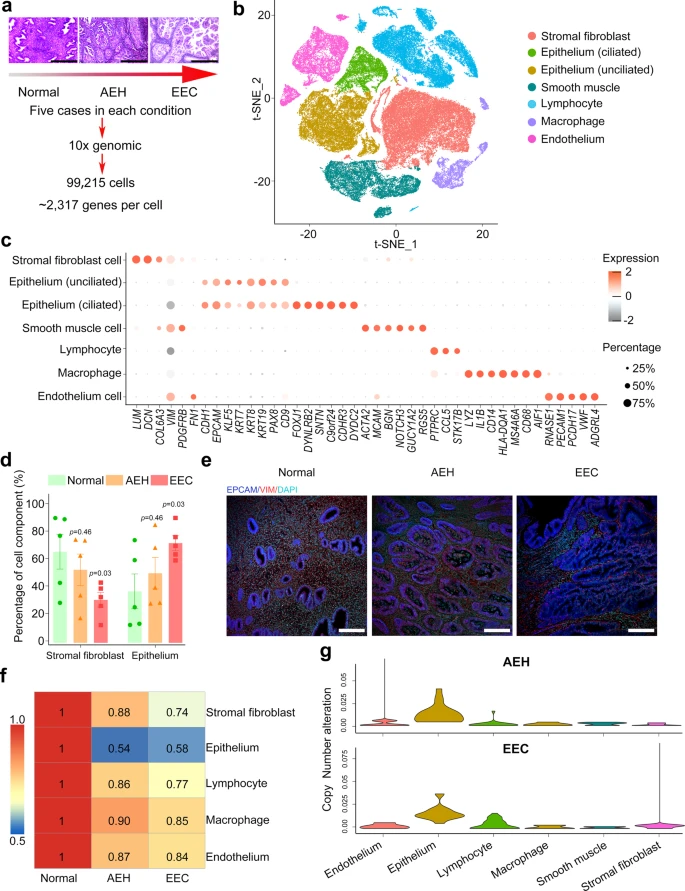
EEC上皮细胞比例和拷贝数变异(CNVs)的增加
比较AEH和EEC的拷贝数与正常的相关性,为下一段EEC的起源提供证据。
- 上皮增生是子宫内膜癌的病理表现,但从正常子宫内膜到AEH和EEC的所有细胞类型比例的动态变化尚未见报道。
- 上皮细胞比例在AEH中增加,在EEC中进一步扩大,而从正常子宫内膜到EEC,间质成纤维细胞比例下降(图1d)并通过组织切片EPCAM(上皮标记物)和VIM(间质标记物)的免疫荧光共染色得到验证。内皮细胞没有变化。
- 细胞群相似性的研究表明,AEH和EEC的上皮细胞与正常组织有很大差异(图1f)。
- 拷贝数变异分析,AEH和EEC的上皮细胞与正常子宫内膜有显著偏差(图1g)。
- 高CNVs通常发生在1号染色体、8号染色体或10号染色体上,与TCGA数据集的结果一致,表明这些可能是可能促进肿瘤进展的典型CNV亚克隆。
EEC起源于子宫内膜上皮细胞,而不是基质细胞
通过CNV相似性,RNA velocity和MET基因分析,表明EEC不来自基质细胞。
- EEC起源假说提示其可能来源于间充质干细胞或子宫内膜上皮干细胞。鉴于上皮细胞比例的增加伴随着间质成纤维细胞比例的减少,作者假设间充质-上皮转化(MET)可能参与了子宫内膜的病理进展。
- 发现不同病理阶段的子宫内膜间质成纤维细胞与正常子宫内膜具有高度的相似性(图1f),不同病理阶段间质成纤维细胞的CNVs较少(图1g)。这些结果表明在间质成纤维细胞中很少发生转录组变化。
- RNA velocity分析,上皮细胞和间质成纤维细胞中的关系,表明MET的谱系轨迹可能不成立。此外,MET调节因子ELF3、OVOL1和OVOL224-26的表达在间质成纤维细胞中几乎检测不到(图2b)。
- 表明EEC很可能来自子宫内膜上皮细胞,而不是间质细胞。

非纤毛腺上皮是EEC来源
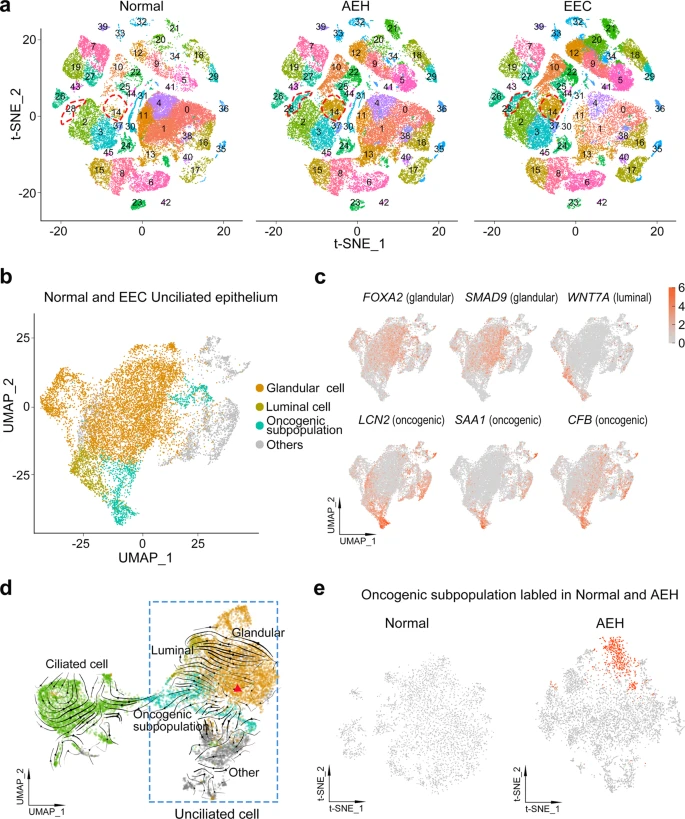
进一步研究上皮细胞,发现AEH和EEC独有的亚群是非纤毛上皮,进一步从非纤毛上皮鉴定出EEC特有的致癌亚群,通过RNA velocity推测出非纤毛上皮腺细胞有可能是致癌亚群的来源。
- 子宫内膜上皮由腺上皮和非腺上皮(也称为腔上皮)组成,两者都含有纤毛上皮和非纤毛上皮。因此,在得出子宫内膜上皮细胞是EEC起源的结论后,作者想进一步研究哪一种子宫内膜上皮是EEC的来源。
- 使用FindClusters研究EEC的上皮亚群,通过无监督聚类根据不同的46个聚类(图3a)。t-SNE显示AEH中存在额外的亚群(聚类14和28)。根据细胞的聚集和特征基因的表达(图1b和补充图2e),是非纤毛上皮细胞,并在EEC样本中持续存在(图3a),这符合前期结果(与正常子宫内膜相比,上皮细胞的相似性较小,AEH和EEC的CNVs较高)
- 在AEH和EEC样本中发现了从非纤毛细胞到纤毛细胞的谱系轨迹(图2a),这表明非纤毛上皮有可能是这些新兴亚群的起源。基于这些结果,坐着推测在AEH和EEC中出现的亚群(14和28)是EEC发育的信号,很可能来自正常的非纤毛上皮。
- 为了进一步研究非纤毛腺上皮,把正常子宫内膜和EEC中的非纤毛腺上皮(EPCAM + / FOXJ1-)筛选出来,进一步聚类,得到26个亚群。几乎所有来自正常样本的细胞都可以被分类为腺细胞(FOXA2, SMAD9)或腔细胞(WNT7A)(图3b, c和补充图5a)。
- EEC样本显示出细胞簇的异质性,除了腺细胞和腔细胞群外,EEC样普遍富集了14、15、26和32亚群(被认为是致癌亚群)。与其他亚群相比,这些亚群的差异表达基因显示出不同的相关通路及特征基因。
- RNA velocity分析来预测单细胞在时间尺度上的状态,以研究EEC上皮的进化轨迹。结果显示非纤毛上皮腺细胞有可能是致癌亚群的来源。
- 使用TransferData鉴定出AEH有致癌亚群,但正常样本中没有。此外,AEH的上皮轨迹显示,非纤毛上皮腺细胞也是推定的致癌亚群的细胞起源。这些结果表明,独特的非纤毛上皮亚群的出现是子宫内膜癌进展的关键步骤,可能对EEC的早期诊断有价值
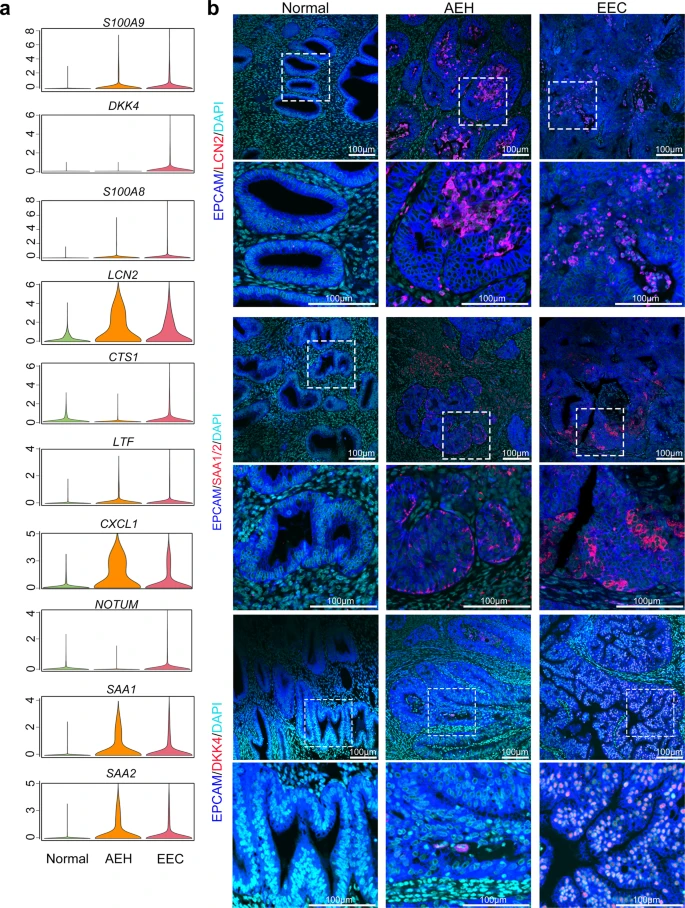
LCN2+/SAA1/2+细胞作是子宫内膜肿瘤发生的一个亚群特征
研究致癌亚群特征基因。
- 为了研究致癌亚群特征基因在人子宫内膜癌病理过程中的规律性,作者比较了正常子宫内膜、AEH和EEC样本中前25个标记基因在非纤毛上皮中的表达水平,发现10个特征基因在EEC中表达显著上调(图4a)。S100A9、S100A8、LCN2、LTF、CXCL1、SAA1和SAA2的上调始于AEH阶段,表明这7个特征基因可能参与了EEC的早期肿瘤发生事件,可作为早期诊断EEC的标志物。
- 此外,不同阶段子宫内膜病变的免疫荧光染色证实,EPCAM+ /LCN2 +细胞出现在AEH和EEC样本中,而不出现在正常子宫内膜中(图4b),这与之前关于LCN2在不同组织学来源的癌症中过表达的报道一致。
- 有报道称SAA(血清淀粉样蛋白A)的产生与肿瘤组织有关,如结直肠癌、卵巢癌、子宫癌、胶质母细胞瘤31和EEC32。然而,SAA是否可以作为EEC早期诊断的标志尚不清楚。作者发现SAA1和SAA2(另一种急性期蛋白)在AEH和EEC非纤毛上皮亚群的表达均增加(图4a),也在免疫荧光染色实验中验证。表明LCN2和SAA1/2可能是子宫内膜早期肿瘤发生的一个特征。
数据库分析确定了子宫内膜癌进展的关键特征基因
联合TCGA进一步分析致癌亚群的特征基因
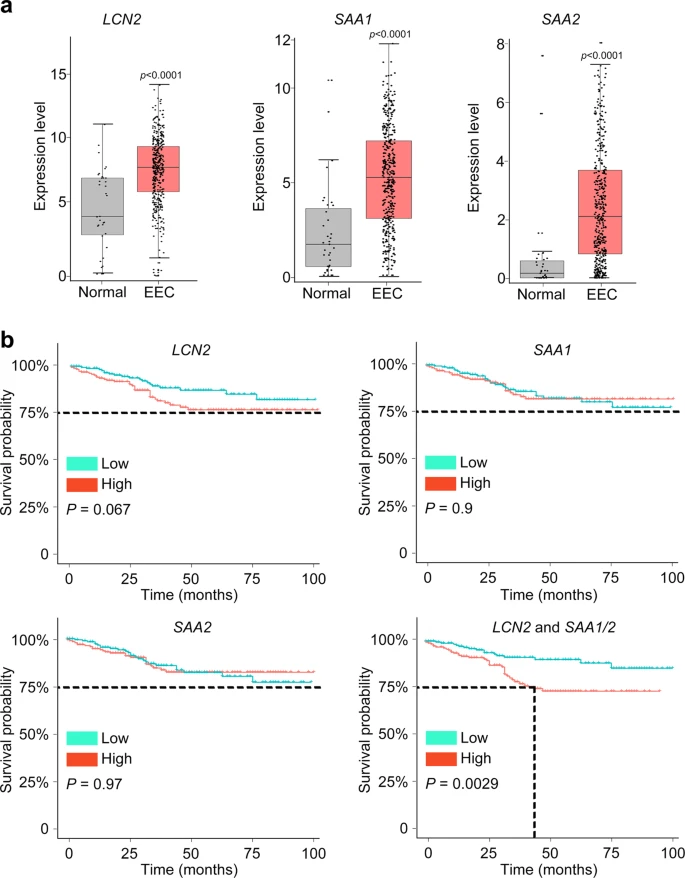
- 为了进一步证实LCN2和SAA1/2表达与子宫内膜癌之间的相关性,分析了TCGA队列,发现上调,并和预后相关。
- 作者还发现了一些候选基因在EEC中特异性表达,如DKK4、CST1和NOTUM,这些基因在EEC中高表达,但在AEH中很少表达(图4a, b)。表明,固有的LCN2 + /SAA1/2+上皮细胞亚群是EEC的早期标志物,DKK4、CST1和NOTUM表达的发展可能是最终癌变阶段的警告信号。
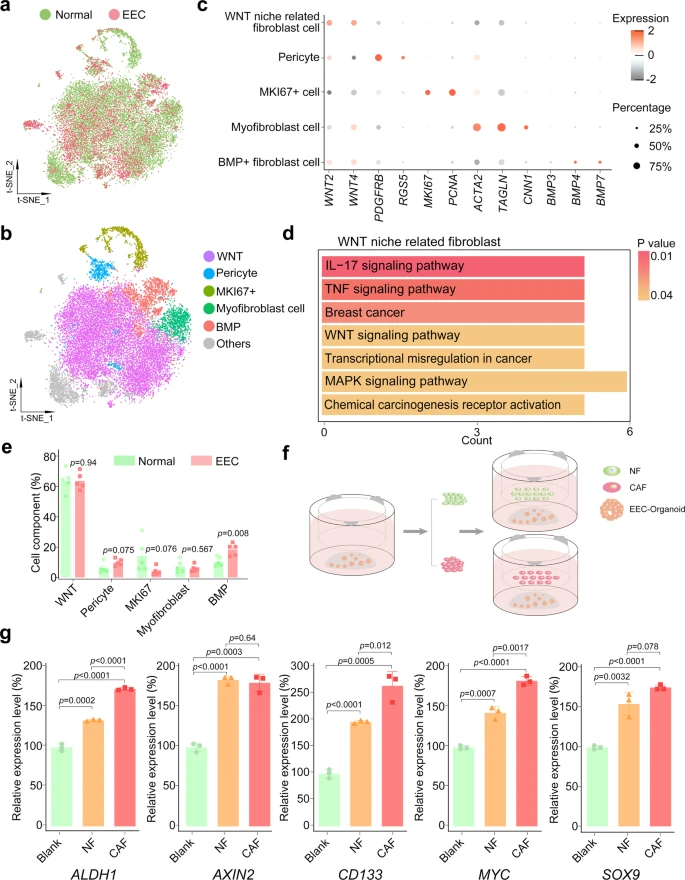
在正常子宫内膜和EEC中,间质成纤维细胞可能是不可缺少的
间质成纤维细胞亚群分析,类器官证明正常子宫内膜的间质成纤维细胞也能促进EEC。
- 肿瘤的进展除了受其内在变化的驱动外,还受到肿瘤微环境的影响。先前的研究表明,正常的基质细胞可能被诱导为癌症相关的间质成纤维细胞(CAFs),从而促进肿瘤生长。因此,对肿瘤发生过程中的间质成纤维细胞进行了表征,以确定间质成纤维细胞如何在EEC中转化,以及它们如何影响肿瘤的进展。
- 与上皮细胞不同,间质成纤维细胞的CNVs和细胞簇的异质性在正常、AEH和EEC之间的转录组差异不显著(图1f、g和附图3c)。t-SNE图显示正常子宫内膜和EEC间质成纤维细胞单细胞有很好的重叠(图6a)。
- 细胞又被进一步分为周细胞(PDGFRB和RGS5阳性)、肌成纤维细胞(ACTA2和TAGLN阳性)和MKI67 +细胞群(图6b)。
- FindMarker功能用于进一步定义其他群体,作者定义了WNT niche相关成纤维细胞(WNT2和wnt4阳性)和BMP+成纤维细胞(BMP3, BMP4和BMP7阳性)亚群(图6b, c)。
- 与正常子宫内膜相对比,细胞类型组成分析显示基质细胞中BMP亚型的比例略有增加,而WNT亚型、周细胞和肌成纤维细胞的比例变化不明显(图6e)。
- 为了证实间质成纤维细胞niche对上皮肿瘤细胞生长的重要性,并研究来自正常子宫内膜的间质成纤维细胞与来自EEC的CAFs之间的差异,使用EEC类器官进行了共培养实验(图6f)。相对表达水平显示,来自正常和癌症供体的间质成纤维细胞促进了EEC类器官中干细胞相关基因(ALDH1、AXIN2、CD133、MYC和SOX9)的上调(图6g)。与来自正常子宫内膜的细胞相比,来自EEC的间质成纤维细胞ALDH1、CD133和MYC的表达更高,这表明EEC中的CAFs支持子宫内膜上皮细胞增殖的能力增强。因此,作者数据表明,在正常子宫内膜和EEC中,所有亚型的间质成纤维细胞可能都是必不可少的。
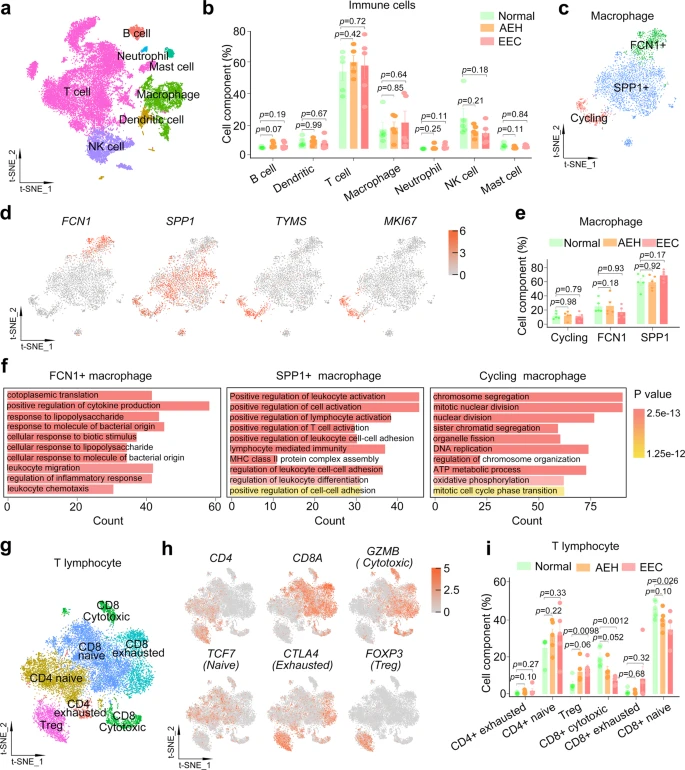
免疫环境的失调可导致子宫内膜肿瘤的发生
免疫细胞亚群关注巨噬细胞和淋巴细胞,分析比例变化。
- 肿瘤相关免疫细胞对TME的建立也至关重要。与正常组织相比,正常、AEH和EEC样本的主要亚型占总免疫细胞的百分比没有明显变化(图7b和附图7d)。
- 进一步研究占免疫细胞很大比例的巨噬细胞和淋巴细胞,分别分析了它们的细胞亚群。
- 巨噬细胞有5个亚群,发现M1 (IL1A和IL1B阳性)和M2 (CD163和IL10阳性)相关基因在相同群体中经常表达(补充图7f)。这一结果挑战了巨噬细胞极化模型(m1型和m2型)。鉴定了巨噬细胞亚群的特征基因,FCN1 + (cluster1), SPP1 + (cluster0, 3,4)和cycling(cluster2, TYMS和MKI67阳性)巨噬细胞(图7c, d),在正常,AEH和EEC样本中,这些亚型在巨噬细胞中的比例没有显著变化(图7e)。
- 最近的研究表明,FCN1 +单核细胞样细胞可以在结肠癌中产生SPP1 +群体,并且在人肺中已经鉴定出以FCN1、SPP1和FABP4表达为标志的三种不同的巨噬细胞亚。提示FCN1和SPP1 可能是巨噬细胞的通用标记物。
- GO分析发现,FCN1 +型巨噬细胞与细胞因子产生和细胞反应的正调节有关,而SPP1 +型巨噬细胞与淋巴细胞活化的正调节有关(图7f)。正如预期的那样,cycling亚型巨噬细胞在细胞周期相关通路中富集(图7f)。
- 们将CD8 T细胞分为naïve (TCF7、IL7R和CCR7阳性)、细胞毒性(GNLY、GZMA、GZMB和GZMH阳性)和衰竭(CTLA4、PDCD1和HAVCR2阳性),CD4 T细胞分为naïve (TCF7、IL7R和CCR7阳性)、衰竭(CTLA4、PDCD1和HAVCR2阳性)和调节性(Treg细胞,FOXP3和TNFRSF4阳性)。
- 与正常样本相比,EEC中细胞毒性和naïve CD8 T细胞的比例显著降低(图7i)。相反,与免疫抑制相关的FOXP3 + CD4 Treg淋巴细胞在EEC样本中显著富集(图7i),提示EEC中免疫逃逸的潜在机制。表明Treg的增加以及细胞毒性和幼稚细胞的减少可能是EEC的特征。
讨论
在单细胞分辨率下全面比较了不同类型子宫内膜细胞在EEC发育过程中的转录特征。我研究描述了子宫内膜病变的动态变化,结果不仅证实了以前报道的重要观察结果,而且为更好地理解EEC的发展提供了信息。
从病理学角度来看,子宫内膜病变的细胞异质性尚未被报道,这严重限制了我们对人类子宫内膜在EEC发育过程中转化的理解。在从正常子宫内膜到EEC的病理进展过程中,除了一般组织学变化外,作者还在单细胞分辨率下分析了不同细胞成分的动态变化,证实了与正常子宫内膜相比,EEC中上皮细胞增加,间质成纤维细胞减少的代表性特征。 通过细胞类型的分类,基于它们的相关性和CNVs分析,发现在EEC的发展过程中,上皮细胞而不是基质细胞或免疫细胞的转录组发生了显著变化。这些发现与EEC作为上皮性癌症的相符合。
结果支持EEC可能起源于非纤毛腺上皮细胞,但仍需要进一步的体内谱系追踪来证实。
通过scRNA-seq鉴定了非纤毛上皮亚群(致癌亚群),这些亚群从AEH阶段开始出现在子宫内膜中,并持续存在于EEC中。通过比较EEC与正常子宫内膜的非纤毛上皮细胞,作者捕获了EEC常见组及其富集途径(TNF、IL-17、Hippo和Wnt信号通路)。在这些途径中,一些已被报道与人类子宫内膜疾病有关。非纤毛上皮亚群的出现可能是EEC的一个重要特征
通过进一步研究致癌亚群的关键标志基因,发现LCN2联合SAA1/2能更好地预测疾病结局。高表达的LCN2与增加的细胞增殖、细胞侵袭和转移有关,并且在EEC中具有侵袭性。同时,据报道SAA是几种癌症的危险因素。但SAA1/ 2在子宫内膜肿瘤发生中的作用尚不清楚。作者发现SAA1/2在AEH中表达升高,在EEC中持续存在,提示SAA1/2表达上调可能是EEC癌前病变的标志。TCGA-EEC数据库对EEC患者预后的分析也证实,LCN2和SAA1/2的高危评分计算与较差的总生存期相关,提示LCN2和SAA1/2有可能作为EEC早期诊断的生物标志物。综上所述,作者得出EEC起源于上皮细胞的结论,而致癌亚群的出现可能是EEC进展的重要指标。然而,这些基因是否是这些亚种群的主要驱动力还有待确定。
目前尚不清楚微环境(包括基质细胞和免疫细胞)如何参与子宫内膜癌的进展,特别是在从正常到AEH和EEC的逐步转变过程中。研究结果与众所周知的现象一致,即在从正常子宫内膜到AEH的过程中,子宫内膜间质细胞的比例减少,在EEC中甚至更低。虽然EEC中基质成纤维细胞的相对数量减少,但体外共培养研究表明,正常子宫内膜和EEC基质细胞均可为EEC类器官的生长提供支持。特别是,CAFs对EEC类器官中干性基因的表达有较强的上调作用,提示间质成纤维细胞的支持作用在肿瘤微环境中可以增强。结果与之前的报道一致,即CAFs促进子宫内膜癌细胞的增殖。尽管如此,为何间质成纤维细胞减少却表现出更强的作用,以及在子宫内膜肿瘤发生过程中正常间质成纤维细胞转化为CAFs的机制尚不清楚。
分析EEC致瘤过程中免疫微环境的动态变化,发现细胞毒性和naïve CD8淋巴细胞群比例下降,CD4 Treg细胞群比例增加,提示子宫内膜发生过程中免疫逃逸。然而,还需要更多的实验验证。
需要指出的是,不太可能获得来自同一患者的材料来研究正常子宫内膜到AEH到EEC的演变,此外研究中scRNAseq的样本量仍然有限。鉴于子宫内膜癌在肿瘤微环境组成和潜在分子改变方面的异质性,本研究结果的解释不能反映所有EEC患者的情况。需要额外的scRNA-seq样本和进一步的体外和体内验证。
同时,分析的子宫内膜癌样本并不能代表所有的子宫内膜癌分子亚型。仅包括染色体不稳定性水平相对较低的样本(NSMP和MMRd),拷贝数高的样本以及POLEmut分子亚型仍有待研究。
总之,作者使用scRNA-seq分析了正常子宫内膜、AEH和EEC的细胞图谱,它们共同代表了EEC的逐步发展。研究表明,EEC起源于非纤毛腺上皮细胞,LCN2 + /SAA1/2+细胞的出现是子宫内膜肿瘤发生的一个特征。最后,作者描述了在EEC进展过程中基质和免疫环境的变化。作者的研究在单细胞分辨率下阐明了EEC发育过程中细胞群的进化和特征,这将有助于子宫内膜癌的研究和诊断策略的发展。
Methods
scRNA-seq data preprocessing
Rawreads in the.fastq files of human endometrial cells were processed in the Cell Ranger Software Suite (10x Genomics Cell Ranger 4.0.0) using refdata-gex-GRCh38-2020-A as reference to map reads on the human genome (GRCh38/hg38), and generated the unique molecular identifier (UMI)matrices. The Cell Ranger outputs were imported into Seurat by the ‘Read10X’ function. Among each sample, cells with UMI counts above upper 10% are removed. Then cells with fewer than 500 UMI counts detected or >40% mitochondrial UMI counts were filtered out. Finally, genes expressed in less than 10 cells were also removed. After the quality filtering, 99,215 cells from 15 subjects were selected for the following analysis.
Unsupervised clustering analysis
The preprocessed data were normalized and scaled using Seurat function NormalizeData, and FindVariableGenes was used to identify highly variable genes. The principal components (PCs) were estimated by RunPCA. UMAP and t-SNE dimensionality reduction were then performed by the RunUMAP and RunTSNE to place cells with similar local neighborhoods in dimension 1 to dimension 40 and visualize the distribution of cells. The cell types were annotated based on the expression pattern of differentially expressed genes (DEGs) and the well-known cellular markers from the literatures. After scaled, we use FindNeighbors with dimensions 1 to dimension 40 to allocated cells. FindClusters was conducted with the parameter ‘res’ to adjust the resolution.
Data integration
Seurat function FindIntegrationAnchors was used to remove batch effect from different batches and integrate single cell data (normalization. method = “LogNormalize”, dims = 1:40, reduction = “cca”).
Marker identification
In order to identify signature genes in each cell type, Seurat function FindMarkers and FindAllMarkers were carried out (min.pct = 0.1, test.use = ‘roc’, return.thresh = 0.25).
Spearman correlation analysis
The expression data was subsetted into different cell types and stages, and the mean expression of cell type markers were calculated to represent the expression level of its corresponding cell type. Finally, the Spearman correlation test was carried out between Normal, AEH and EEC.
Estimation of CNVs
The R package InferCNV was used to identify somatic large-scale chromosomal copy number variation of each gene (cutoff = 0.1), using the control group as reference60,61. Heatmaps were thenmade to show copy number variation with specific cell type and segments of chromosome. In order to inspect the copy number alteration fromdifferent cell types, separately, we adjusted the HMM prediction matrix to zerocentered, and calculated the sum of square to get cell-based level of copy number variation.
Calculation of RNA velocity
We performed the RNA velocity analysis using the Python package scVelo to inform the transition among cell types. The BAM file from Cell Ranger were used to generate count matrices of pre-mature (unspliced) and mature (spliced) abundances, using the ‘velocyto’ package62, and the latter were used as the input for scVelo afterwards. The dynamical model in scVelo was used, and the results were projected back to UMAP generated by Seurat.
Pseudobulk RNA-seq based PCA and gene enrichment analysis
For PCA, we performed pseudobulk RNA-seq analysis: The UMI counts from cells of each sample were summed up, generating a pseudobulk RNA-seq expression matrix. The matrix was log2 transformed and exported for PCA analysis using plotPCA from DESeq2 package afterwards. GO and KEGG enrichment analysis were carried out using enrichGO and enrichKEGG using the R package clusterProfiler.
文章作者 zzx
上次更新 2024-01-12