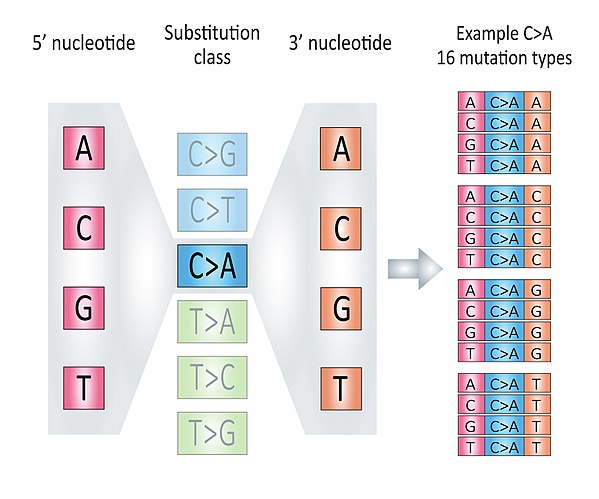
拿到突变的数据之后,一般会先看除了进行突变注释,看突变所在的基因和造成的氨基酸变化,还会看具体的碱基变化类型,共96种,以三个碱基为统计单位,看三联核苷酸中间的碱基变化类型的个数,以C变为A为例(等同负链G变为T),NCN>NAN,N可以为任意碱基,所以有4*4=16种,所有情况为96种。
统计96种突变类型的具体数目,之后会进行突变的signature分析有三种:
- 与已知的cosmic signature进行比较,看哪些signature比重高
- 利用非负矩阵分解NFM找novel的signature
- 分析1和2之后,看novel的signature和已知的signature的相似性
突变的signature是内外部环境共同作用下造成的一些特定的变异特征,这些因素包括DNA修复或者复制缺陷,吸烟饮酒等等。
找cosmic的signature
利用R包deconstructSigs进行
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
|
# 假设你有一个数据框,包含了突变的信息,以包自带的sample.mut.ref为例
head(sample.mut.ref)
# Convert to deconstructSigs input,得到96碱基突变数据框
# 指定染色体位置、突变碱基和样本名所在的列名
sigs.input <- mut.to.sigs.input(mut.ref = sample.mut.ref,
sample.id = "Sample",
chr = "chr",
pos = "pos",
ref = "ref",
alt = "alt")
# 提取样本的mutation signature
# 这里的signature.nature2013或者signatures.cosmic就是参考的signature,如果你有自己的signature数据,比如SomaticSignatures包找到的,也可以在这个地方指定,这样就计算的是提供的singature
sample_1 = whichSignatures(tumor.ref = sigs.input,
signatures.ref = signatures.nature2013, # 参考signature
sample.id = 1, # 样本的barcode
contexts.needed = TRUE,
tri.counts.method = 'default' # 是否进行normalzie
)
sample_1$weights[1:3]
# Signature.1A Signature.1B Signature.2
#1 0 0.1564832 0
|
找novel的signature
利用SomaticSignatures包进行,这里找到的novel signature可以作为参考的signature用deconstructSigs,注意96突变类型数据框的名应一致。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
|
library(SomaticSignatures)
library(SomaticCancerAlterations)
library(BSgenome.Hsapiens.1000genomes.hs37d5)
sca_metadata = scaMetadata()
# sca_data = unlist(scaLoadDatasets()) # 官网用的多种是癌症的数据
# 我们以TCGA的LUAD的全外数据为例
sca_data = (scaLoadDatasets())$luad_tcga
sca_data$study = factor(gsub("(.*)_(.*)", "\\1", toupper(names(sca_data))))
sca_data = unname(subset(sca_data, Variant_Type %in% "SNP"))
sca_data = keepSeqlevels(sca_data, hsAutosomes(), pruning.mode = "coarse")
# 构建对象,这里可以自己根据自己的数据构建VRanges对象
sca_vr = VRanges(
seqnames = seqnames(sca_data),
ranges = ranges(sca_data),
ref = sca_data$Reference_Allele,
alt = sca_data$Tumor_Seq_Allele2,
sampleNames = sca_data$Patient_ID,
seqinfo = seqinfo(sca_data),
study = sca_data$study)
sca_motifs = mutationContext(sca_vr, BSgenome.Hsapiens.1000genomes.hs37d5)
# 这里得到96碱基突变数据框
sca_mm = motifMatrix(sca_motifs, normalize = TRUE)
# 可视化
plotMutationSpectrum(sca_motifs)
# 找最佳的signature数目,一种是NMF,一种是PCA方法
n_sigs = 2:8
gof_nmf = assessNumberSignatures(sca_mm, n_sigs, nReplicates = 5)
gof_pca = assessNumberSignatures(sca_mm, n_sigs, pcaDecomposition)
# 确定最佳的数目
plotNumberSignatures(gof_nmf)
plotNumberSignatures(gof_pca)
# 找signature
n_sigs = 5 # 假设最佳的是5
sigs_nmf = identifySignatures(sca_mm, n_sigs, nmfDecomposition)
sigs_pca = identifySignatures(sca_mm, n_sigs, pcaDecomposition)
library(ggplot2)
# 看每个样本的signature的权重
plotSignatureMap(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Heatmap")
# 看每个样本具体的突变类型统计
plotSignatures(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Barchart")
# 没放图,具体的图可以参考 https://bioconductor.org/packages/release/bioc/vignettes/SomaticSignatures/inst/doc/SomaticSignatures-vignette.html
|
另外还可以通过cosine similarity 比较两个signature之间或者样本之间的相似性。
参考
https://github.com/raerose01/deconstructSigs
https://bioconductor.org/packages/release/bioc/vignettes/SomaticSignatures/inst/doc/SomaticSignatures-vignette.html
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
#####################################################################
