随机森林训练集样例不平衡
最近在用RF的时候,有一个很明显的现象就是训练集样本标签不平衡,有的很多有的很少,导致做预测的时候,预测的标签倾向于在训练集中占多数的标签。

机器学习的目标是做预测,但有时候单个预测模型并不一定有很好性能。集成学习的思想是,把多个弱机器学习模型集成在一起,不同的学习方法之间可能相互补充,进而降低预测的错误率,提高最终的预测性能。集成学习在各个规模的数据集上都有很好的策略结果。
常见的集成方法有Bagging,Boosting,Stacking,Voting和Blending。
又可以分为两大类:(1)序列集成方法:其中参与训练的基础学习器按照顺序生成(Boost)。序列方法的原理是利用基础学习器之间的依赖关系。通过对之前训练中错误标记的样本赋值较高的权重,可以提高整体的预测效果。(2)并行集成方法,其中参与训练的基础学习器并行生成(如经典的Random Forest)。并行方法的原理是利用基础学习器之间的独立性,通过平均可以显著降低错误。
CCTB 2022:https://biomarker2022.sciconf.cn
参加肿瘤标志物的大会,好几个会议同步进行,来演讲的人都是业界的专家,水平很高,虽然大部分报告都是科研形式的汇报,和产业汇报不一样,但同样给人启发。

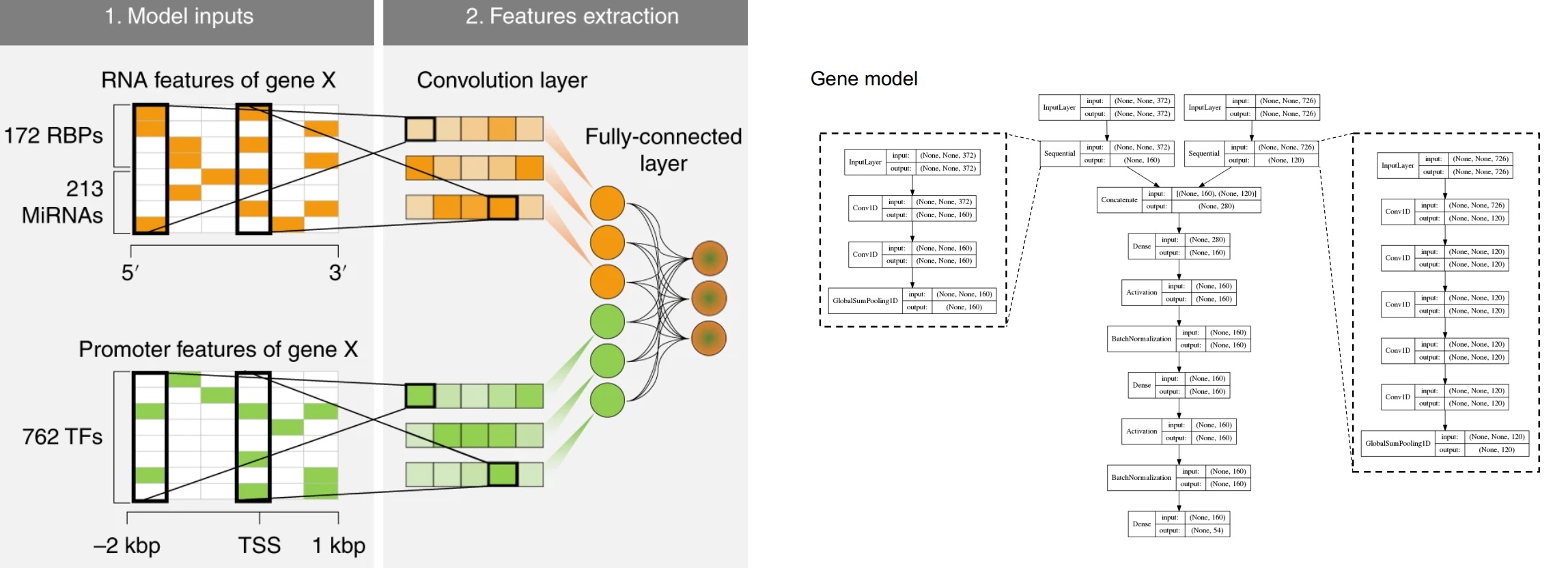
保存几张1D卷积的图和文章,方便以后查找,摘自互联网。说不准以后搞深度学呢🫠🙃卷的结果就是以后不上人工智能都不要意思说自己是搞生信的。
主要是看到一篇文章,用了卷积神经网络,我一直就好奇组学数据怎么做卷积,所以就看了下文章,发现用的是Conv1D。(不知道理解的对不对)对于1D卷积Conv1D而言,如果卷积核kernel size是2的话,最终会生成一个 “行数-kernel size+1“的向量,如果数据分批给的话,就有batch,比如如果样本是21000,batch size是128的话,每个batch有165个样本,所以Nature Machine Intelligence的附图 Fig. 10这篇文章还进行了BatchNormalization。
Samples = 21000,batch_size=128-> training_sample for each epoch =21000/128 = 164.06 ~= 165
filters可以指定多次卷积(相同kernel size),这样可以生成二维的数据。
| Args | https://www.tensorflow.org/api_docs/python/tf/keras/layers/Conv1D |
|---|---|
filters |
Integer, the dimensionality of the output space (i.e. the number of output filters in the convolution). |
kernel_size |
An integer or tuple/list of a single integer, specifying the length of the 1D convolution window. |
Deep learning decodes the principles of differential gene expression
分析的代码已经调试好,但分析的时间较长;
后端启动jupyter notebook后,奈何网络不稳定,notebook经常掉线,跑到一半的程序就断掉了;
服务器其他人的jupyter notebook的端口如果和我的一样,别人启动jupyter notebook后,我正在用的端口就会往后变。
于是我就想,能否在终端直接运行.ipynb文件,这样我就可以加nohup命令了,或者把ipynb的代码转成python,我nohup运行python也行。
基于以上情况,我google到了nbconvert。
nbconvert的github地址:https://github.com/jupyter/nbconvert
jupyter nbconvert通过模版引擎jinja将ipynb文件转成其他格式的文件,包括
此外nbconvert还有另外一个功能就是通过–execute选项在终端执行ipynb文件
|
|
|
|
上面是格式转换,在转换的过程中,比如转pdf、latex的时候,可能还需要额外的包,比如pandoc等,还需要额外安装。可以参考https://nbconvert.readthedocs.io/en/latest/index.html
jupyter nbconvert还有个功能就是执行ipynb格式的文件,如下
|
|
假设我想把通过运行jupyter nbconvert执行ipynb文件的过程更简单点,可以通过在.profile里面设置命令的别名
|
|