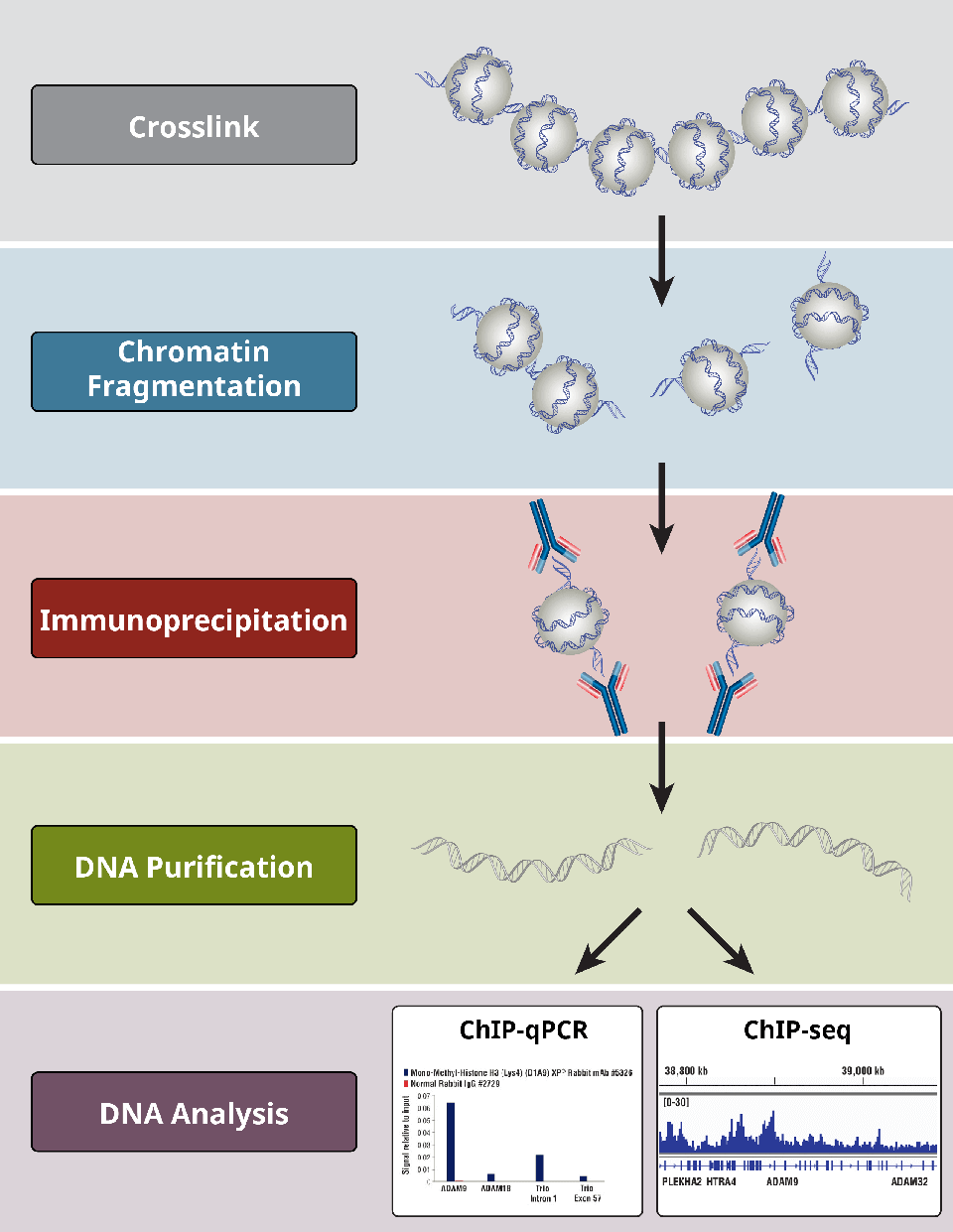
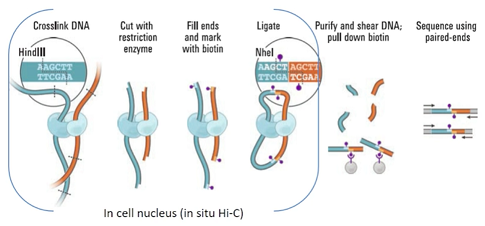
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
|
String encoding mismatched and deleted reference bases, used in conjunction with the CIGAR and SEQ fields to reconstruct the bases of the reference sequence interval to which the alignment has been mapped. This can enable variant calling without requiring access to the entire original reference.
编码错配或del的字符串,与CIGAR和SEQ一起使用,重建read的比对情况。可以在不需要整个参考序列的情况下,用于突变检测。
The MD string consists of the following items, concatenated without additional delimiter characters:
MD字符串由以下不含分隔符的条目组成。
• [0-9]+, indicating a run of reference bases that are identical to the corresponding SEQ bases;表示与相应SEQ碱基相同的一系列参考碱基;这里[0-9]+是正则表示。
• [A-Z], identifying a single reference base that differs from the SEQ base aligned at that position;在这个位置SEQ碱基与参考碱基不一致
• \^[A-Z]+, identifying a run of reference bases that have been deleted in the alignment.以^开头,表明有个del,注意del前有^。
总结:数字表示匹配,碱基表示错配,^碱基表示del。
As shown in the complete regular expression above, numbers alternate with the other items. Thus if two mismatches or deletions are adjacent without a run of identical bases between them, a ‘0’ (indicating a 0-length run) must be used to separate them in the MD string. 数字和字符交替出现,如果两个连续错配,中间需要用0来隔开。
Clipping, padding, reference skips, and insertions (‘H’, ‘S’, ‘P’, ‘N’, and ‘I’ CIGAR operations) are not represented in the MD string. When reconstructing the reference sequence, inserted and soft-clipped SEQ bases are omitted as determined by tracking ‘I’ and ‘S’ operations in the CIGAR string. (If the CIGAR string contains ‘N’ operations, then the corresponding skipped parts of the reference sequence cannot be reconstructed.)
Clipping, padding, reference skips, and insertions (‘H’, ‘S’, ‘P’, ‘N’, and ‘I’ CIGAR operations)不体现在MD字符串中,所以要与CIGAR结合。
For example, a string ‘10A5^AC6’ means from the leftmost reference base in the alignment, there are 10 matches followed by an A on the reference which is different from the aligned read base; the next 5 reference bases are matches followed by a 2bp deletion from the reference; the deleted sequence is AC; the last 6 bases are matches.
10A5^AC6表示10个匹配(10),一个与A不匹配(A),2bp的del(^AC),6碱基的匹配。
|