
VCF文件中的原始突变过滤--filter raw variants in vcf
文章目录
Hard filter突变的传统过滤方式
此时VCF文件中的突变,与刚开始下机得到的FASTQ文件类似,称为raw data。此时的突变集合中,有很多假阳性突变,这些突变需要在突变分析之前过滤掉。
传统的过滤方式,直接根据每个突变的注释信息,进行过滤。最直接和最常见的是根据DP标签过滤,即根据该突变位点的测序深度进行过滤。通常,深度越低,支持该突变的reads数目越少,该突变越不可信。还可以根据前面提到的QUAL质量分值进行过滤,分值越低越不可信。Forward reads和Reverse reads的比例。通过,设定一定的阈值,看这些注释信息是高于还是低于该阈值。
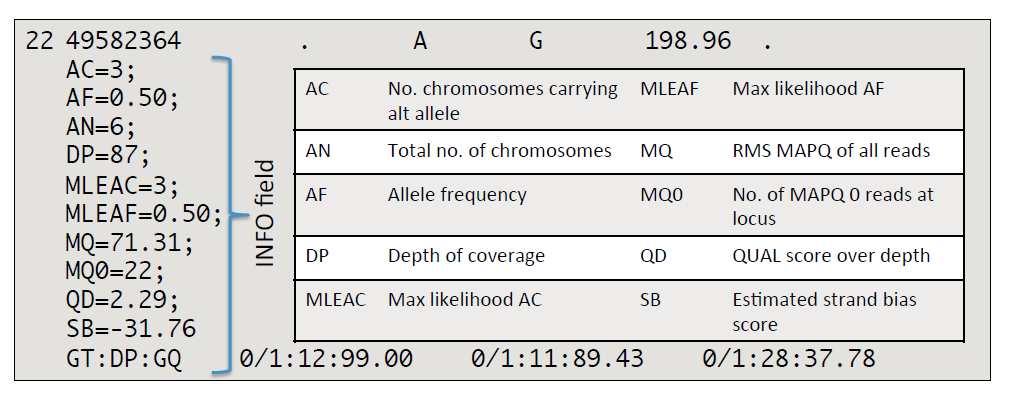
这种直接根据突变信息进行过滤的方式,GATK成为hard filter。GATK常用的hard filter参数还有DP < 10,QD < 2.0,FS > 60.0,MQ < 10.0,MQRankSum < -12.5,ReadPosRankSum < -8.0等方式。这些阈值通过GATK的VariantFiltration工具进行过滤,突变满足其中一条,就会被过滤。
这种过滤方式直接根据特定阈值就将突变过滤掉,考虑的维度较少,真实突变可能因为某一注释没有到达阈值而被错误的过滤掉。如果为了保留这些真实的突变,而放松阈值,又可能同样将假阳性突变保留。
Hard filter常用于panel测序,panel测序得到的突变位点较少,不足以通过机器学习的方式进行过滤。
VQSR突变质量分值再校准
虽然这步叫做Varaint Quality Score Recalibrate,但该步并没有再校准突变质量分值,而是重新生成了一个叫做VQSLOD (for variant quality score log-odds)的分值,且认为该分值是被充分校准的。
VQSR不同于传统的hard filter。VQSR整合突变的多种维度的信息,如下图,通过机器学习的方式,得到好的突变good variant和坏的突变bad variant的特征范式,对突变进行过滤,从而挑选出聚簇在一起的好的突变。需要注意的是,VQSR只利用INFO的注释信息,而没有利用样本特异性的SAMPLE信息。
通常用于训练的数据集,是通过分析得到的raw variant data数据集与hapmap、omni、1000Genome等(hapma和omni中的突变通常被认为是高质量的、验证过的,1000 Genome中的突变可信度次之)进行比较,得到raw variant data数据集中高可信的突变集合,集合中的突变注释信息将被用于机器学习训练模型。同理,低质量假阳性突变的数据也同样。通过机器学习,会对训练数据集高可信的突变的分布进行建模和聚类。同时,也对所有突变进行建模,计算VQSLOD值。通常离聚类形成的簇核心近的突变,VQSLOD值越高,越可信。
在最终过滤突变的时候,GATK VQSR并没有一个直接的阈值进行过滤,而是通过引入sensitivity灵敏度的概念。比如过滤的阈值是99%,则意味着训练数据集中有99%的突变也位于hapmap中,此时的VQSLOD值就是用于突变过滤的阈值。VQSLOD值高于该阈值的突变会被标为PASS,低于的则被过滤,并在FILTER列标记。 因为SNP和INDEL的特征不同,VQSR通常将SNP和INDEL分开进行过滤。
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2018-05-21