
文件介绍--FASTQ文件格式
文章目录
在培训部门同事的时候,发现刚开始学生信的人,只是在学如何运行命令,但对自己手头的文件格式和内容却不了解,这对分析的流程的深入理解和研究是非常不好的,所以刚学习的人,应该在等待分析结果的时候,多去了解下文件的内容,程序的大体算法等,这对以后的工作优化是非常有好处的。本文简单介绍一下Fastq的文件格式,希望新手多查文档,多了解自己接触的东西。
文库构建和测序
DNA分子会通过超声波或者酶被打断成几百碱基的小片段,然后在小片段DNA分子的两端添加接头,便于测序和样本区分。当然现在也有转座酶技术,通过转座酶同时实现DNA片断化和加接头和引物的过程。将文库上机,文库中的DNA分子首先与flowcell上lane中的接头结合,通过桥式PCR进行扩增(cluster簇增长),待达到一定量之后,进行便合成边测序。
测序数据的产生
每进行一个cycle,测序仪会合成过程中产生的荧光进行拍照分析,将产生的数据记录在bcl(base calling)文件,测序仪后续会将bcl文件转化成fastq文件,并同时进行demultiplexing。Demultiplexing会将属于同一Index的reads放在同一个fastq文件中,就个过程是Illumina测序仪自动化进行的,如果涉及更复杂参数的demultiplexing,可以下载illumima的bcl2fastq程序,指定相关文件夹即可自己进行demultiplexing。因为在测序上机之前,会在测序仪中设置样本对应的index,这样通过demultiplexing,测序仪对每个样本产生一对fastq文件。Illumina测序仪会自动将拍照形成的bcl文件转换成FASTQ文件。此文件为数据分析的开始。
FASTQ文件格式简介
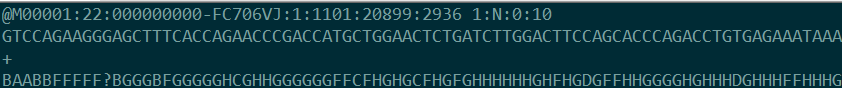
Paired End(PE)测序,会生成的一对FASTQ文件,分别为R1和R2,正是因为双端测序的产生,使得序列拼接和比对更加准确,因为如果单端的话,只能依靠单端read的长短信息,如果双端的话,能依靠整个插入DNA片断长度的信息,通过判断配对序列的相近距离,确认reads的位置,减少了read比对到多个地方的情况发生。 配对的reads分别在两个FASTQ文件中,其中一个FASTQ文件的内容如下,每四行表示一条read:
|
|
• 第一行以@字符开头,后面跟测序仪的编号和其他描述信息。
|
|
| M00001 | 测序仪设备编号 |
|---|---|
| 22 | the run id |
| FC706VJ | the flowcell id |
| 1 | the flowcell lane |
| 1101 | tile number within the flowcell lane |
| 20899 | ’x’-coordinate of the cluster within the tile |
| 2936 | ’y’-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| N | Y if the read is filtered, N otherwise |
| 0 | 0 when none of the control bits are on, otherwise it is an even number |
| 10 | a sample number (as taken from the sample sheet) in place of an index sequenc |
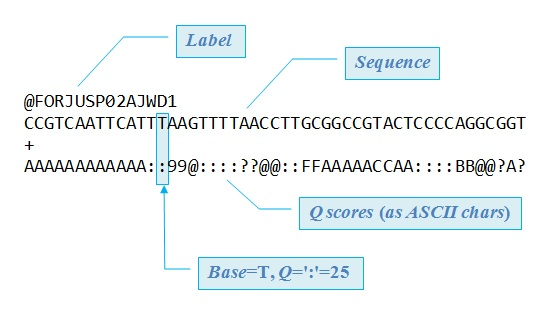
• 第二行为测序的原始序列 • 第三行一般为字符+ • 第四行表示第二行序列中每个碱基的质量,即每个碱基的可靠性。因为每个碱基都有一个质量,所有第四行的字符数与第二行的字符数一样多。质量又与Phred score有关。后续的序列预处理和比对会参考测序碱基的Phred质量分值。 假设第二行的一个碱基在测序过程中被识别错误的概率为e(由测序仪计算产生),则该碱基的质量即Phred score的可以表示为Q_phred=-10〖log〗_10 e,即e取对数,乘以-10。错误率越低,碱基的Phred score质量值越高。IlIumina最新的现在的分值范围为0到41。 碱基的Phred质量分值与错误率的对应关系为。
| Phred Quality Score | Probability of incorrect base call | Base call accuracy |
|---|---|---|
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.9% |
| 40 | 1 in 10,000 | 99.99% |
该碱基的Phred质量分值还需要换算成FASTQ中第四行的字符。将碱基的Phred质量分值加上33,得到的数换成对应的ASCII编码字符,即得到第四行中对应的字符。对应关系如下。

此时又叫做Phred+33,以前还有Phred+64,即将phred分值加上64后换成对应的ASCII编码字符。
#####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: 感谢好友SYY&33,& Jason
#####################################################################
文章作者 zzx
上次更新 2018-05-08