
GATK Best Practices:通过GATK4 docker运行processing-for-variant-discovery-gatk4.wdl
文章目录
Run GATK Best Practices for data pre-processing by Cromwell/WDL
与GATK4正式发布的还有WDL(workflow description langaue,https://software.broadinstitute.org/wdl/),WDL将工作流程分为了workflow, task, call, command 和 output。
与以往GATK提供Best practice的PPT介绍不同,现在Broad提供的是Best practice(https://software.broadinstitute.org/gatk/best-practices/)的WDL文件。WDL文件运行通过cromwell运行,并且有json格式的参数输入文件指定WDL文件中流程所需要的参数。比如
sudo java -jar cromwell.jar run workflow.wdl --inputs workflow.inputs.json
我们只需要修改json文件中的参数就可以运行gatk4 Best Practices,而不需要自己去搭建流程,简化了工作量,也遵循了Broad提供的推荐设置和流程。本文只介绍突变检测前的序列比对和recalibrate这部分的GATK best practices,该流程生成了用于variant calling的bam文件。
1,文件准备
WDL文件和json文件 Broad在github上提供了进行突变检测call variant之前的数据处理data proceesing流程,见https://github.com/gatk-workflows/gatk4-data-processing 从github上,我们需要下载两个文件 processing-for-variant-discovery-gatk4.wdl (用于data pre-processing 的 pipeline) processing-for-variant-discovery-gatk4.hg38.wgs.inputs.json(指定WDL的参数文件)
ubam文件: 要求ubam文件中要有RG tag,经过排序sort之后,该文件可以通过picard将fastq文件转换得到
GATK resoure bundle,从中下载GATK需要的dbsnp文件,known site等文件 ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/ https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0/
2, 软件准备
下载cromwell,用于运行WDL 官网:http://cromwell.readthedocs.io/en/develop/tutorials/FiveMinuteIntro/
下载GATK docker镜像,根据json文件中的第5部分DOCKERS,还需要下载python和genomes in the cloud 镜像,pull命令中加入registry.docker-cn.com,可以指定docker从国内源中下载,避免下载速度较慢或不能下载。
|
|
3,修改json文件
json文件中的gs://路径下的文件是google storage cloud上面的文件,可以换成本地电脑的路径。
 第一部分 SAMPLE NAME AND UNMAPPED BAMS:
该部分指定样本名和分析需要的起始ubam文件,该部分会经常修改
第一部分 SAMPLE NAME AND UNMAPPED BAMS:
该部分指定样本名和分析需要的起始ubam文件,该部分会经常修改
| sample_name | 样本名 |
|---|---|
| ref_name | 基因组版本 |
| flowcell_unmapped_bams_list | ubam的list文件路径,该路径中每一行为一个ubam的绝对路径 |
| unmapped_bam_suffix | ubam文件后缀 |
第二部分 REFERENCE FILES:
修改成参考基因组的fastq文件、dict文件和用bwa index之后生成的alt、ann、bwt文件等路径。该部分只需要修改一次,后续不用在修改。
第三部分 KNOWN SITES RESOURCES:
从GATK resource bundle中下载的文件,按照要求改成本地路径即可。

第四部分 MISC PARAMETERS
不用修改。因为我们下载的docker镜像中含有预先编译好的gatk,bwa,picard等程序
第五部分 DOCKERS
如果不确定,可以通过sudo docker images查看 REPOSITORY TAG IMAGE ID CREATED VIRTUAL SIZE registry.docker-cn.com/library/python 2.7 b1b87432bf0e 42 hours ago 678.2 MB registry.docker-cn.com/broadinstitute/gatk latest 5e9cb2ded24b 2 weeks ago 4.882 GB registry.docker-cn.com/broadinstitute/genomes-in-the-cloud 2.3.1-1512499786 989127e46444 3 months ago 1.879 GB 在json文件中将对应的docker镜像改成本地有的,切记tag要写对。
第六部分 PATHS

不用修改,broadinstitute/genomes-in-the-cloud的iamge镜像生成容器中的路径, 如果使用sudo docker run -it 进入broadinstitute/genomes-in-the-cloud的话,可以看到/usr/gitc/路径中的其他文件 同样,/gatk/gatk是broadinstitute/gatk的image镜像生成的容器中的路径,所以不用修改。
第七到十部分也不用修改。
因为cromwell运行WDL的时候会自动调用docker,所以我们不需要关注docker怎么使用,甚至不用关系流程,因为都是broad提供,我们只需提供json文件即可。如果想在docker上跑gatk4,可以参考https://gatkforums.broadinstitute.org/gatk/discussion/10172/how-to-run-the-gatk4-docker-locally-and-take-a-look-inside
4,运行GATK4 Best practices中的data preprocessing流程
|
|
刚开始时会生成一个workflow的id,中间文件都会在该文件夹内。
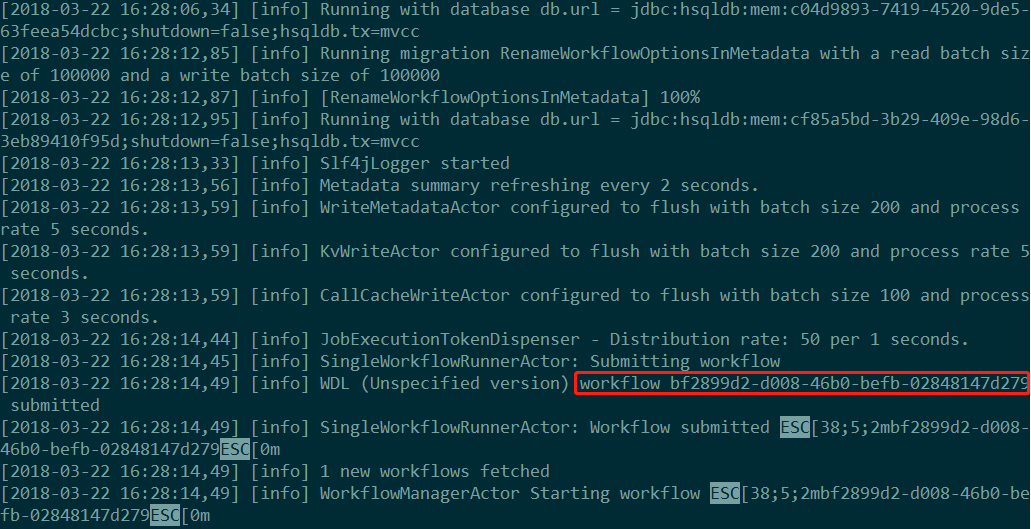
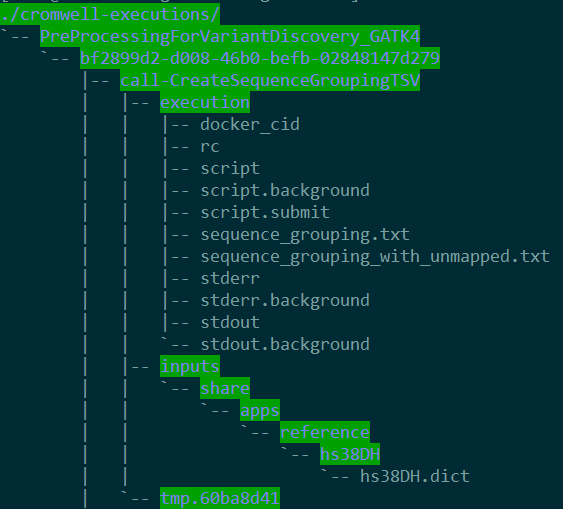
如果中间运行出错,可以查看workflow文件夹中的stderr文件。cromwell的文件夹结构如下

运行结束后,会收到 [2018-03-22 19:04:55,04] [info] SingleWorkflowRunnerActor workflow finished with status ‘Succeeded’.的提示。
就这样,我在不了解docker的情况下,在没有关注GATK Best Practices具体流程的情况下,对ubam进行比对,mark duplicate,recalibrate等操作生成了用于variant calling的bam文件。具体的Best practices流程,需要通过研究processing-for-variant-discovery-gatk4.wdl文件。后续在分析其他样本时,只需要将json文件中的第一部分修改即可。
下一步就是通过WDL进行germline variant calling的工作
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2018-03-22