
样本突变频谱分析和基于突变频谱的热图聚类
文章目录
对突变频谱(Mutation spectrum)进行分析,可以得知样本各种类型突变(如C:G>T:A)的数量及样本是否具有某种类型突变的偏好性。
单个碱基的替换一共有六种变异类型:C>A/G>T,C>G/G>C,C>T/G>A,T>A/A>T,T>C/A>G,T>G/A>C,有可以根据发生替换的碱基类别分为两大类:颠换transversion,嘌呤与嘧啶之间的替换,转换transition,嘌呤与嘌呤或嘧啶与嘧啶之间的替换。根据各类型点突变的比例,对样本和点突变类型进行聚类分析,可以研究样本点突变类型的偏好和各样本在点突变水平上的相似程度。
1,突变频谱数据准备
从VCF或者Annovar注释之后文件中,提取突变类型,第一列为样本名,第二列为突变类型,命名为type.txt。有来自来个实验的10个样本,分别为S1–S7和CHG。
|
|
2,生成突变频谱图
|
|
用到了ggplot2的geom_bar,生成图片如下,横坐标为样本,纵坐标是不同类型的突变所占的比例,可以看出CHG样本的T>C/A>G类型的突变频率要比S样本低,T>A/A>T类型的突变频率较S样本要高。
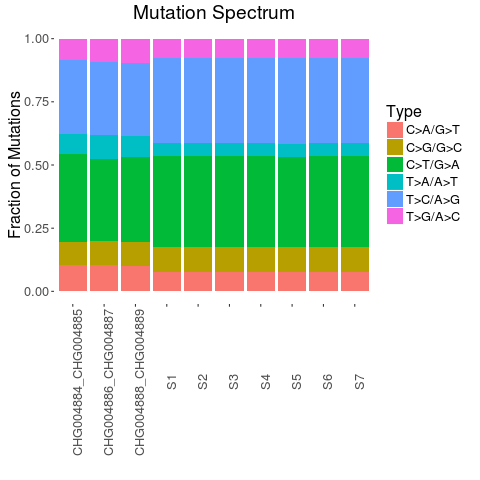
还有一种突变频谱,就是考虑突变位点的两旁的各1bp的碱基,组成464共96中类型突变,会更加详细,如果有机会,我会在后续介绍。
3,热图数据准备
根据type.txt样本,统计每个样本每种类型突变所占的比例,存在名为ratio.txt的文件中,如下,只显示前三行:
|
|
4,绘制热图和聚类
|
|
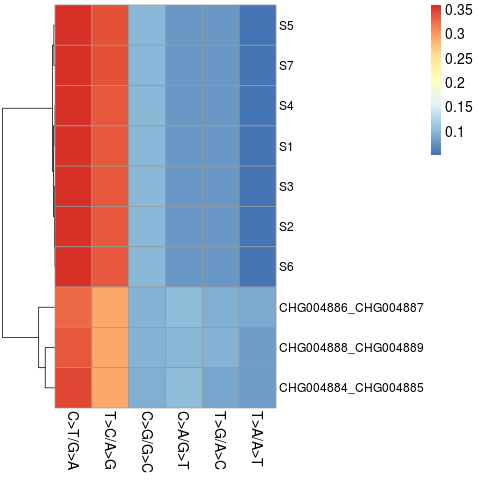
结果如下图,从深红到深蓝表示突变频率由大到小,每一行代表一个样本,每一列代表一种突变类型,可以看到每个样本的突变频率热图,并且S实验和CHG实验的样本分别聚成了一类,被区分出来。
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
###################################################################
文章作者 zzx
上次更新 2016-04-03