
Paired end sequencing VS Mate pair sequencing
文章目录
Mate pair sequencing
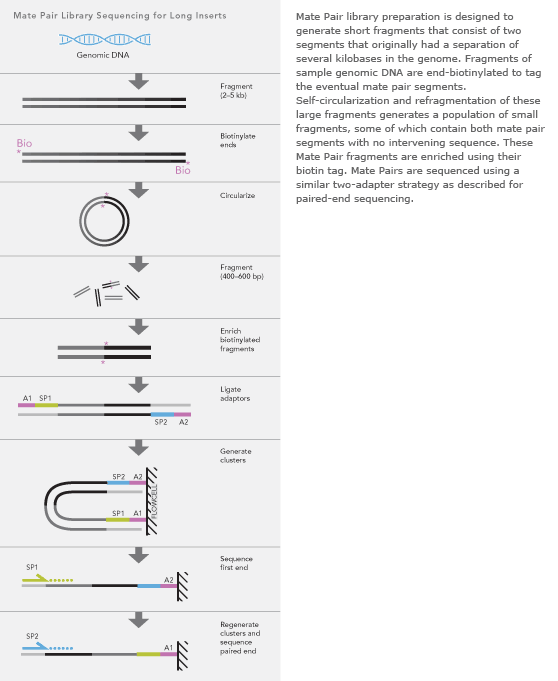
Mate Pair Library Sequencing enables the generation of libraries with inserts from 2 to 5 kb in size. These long-insert Paired-End libraries are useful for a number of applications, including De NovoSequencing, genome finishing, and structural variant detection.
Combining data generated from Mate Pair library sequencing with that from short-insert paired-end reads provides a powerful combination of read lengths for maximal genomic sequencing coverage across the genome. Following DNA fragmentation, 2-5 Kb fragments are end-repaired with biotin labeled dNTPs. The DNA fragments are circularized, and non-circularized DNA is removed by digestion. Circular DNA is fragmented and fragments biotin labels (corresponding to the ends of the original DNA ligated together) are affinity purified. Purified fragments are end-repaired and ligated to Illumina Paired-End sequencing adapters. Additional sequences complementary to the flow cell oligonucleotides are added to the adapter sequence with tailed PCR primers. The final prepared libraries consist of short fragments made up of two DNA segments that were originally separated by several kilobases. These libraries are ready for paired-end cluster generation followed by paired-end sequencing utilizing the Genome AnalyzerIIx system. Highlights High Genomic Diversity: Efficient protocol enables the highest genomic diversity of any next-generation platform User-Friendly Workflow: Simple two-and-a-half day workflow with limited hands-on time and multiple stopping points Low DNA Input Requirements: Requires as little as 10 μg of starting material.
Paired end sequencing
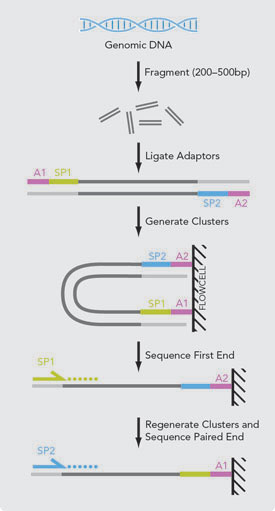
A simple modification to the standard single-read DNA library preparation facilitates reading both the forward and reverse template strands of each cluster during one paired-end read. In addition to sequence information, both reads contain long range positional information, allowing for highly precise alignment of reads. The Paired-End Sequencing Assay utilizes a combination of cBot (or the Cluster Station) and the Paired-End Module followed by paired-end sequencing on the Genome AnalyzerIIx. The unique paired-end sequencing protocol allows the end user to choose the length of the insert (200–500 bp) and sequence either end of the insert, generating highly quality, alignable sequence data. A typical paired-end run can achieve 2 × 75 bp reads and up to 200 million reads. Paired-End Sequencing Highlights Simple Paired-End Libraries: Simple protocol workflow allows generation of unique ranges of insert sizes Efficient DNA Use: Requires the same amount of DNA as single-read, 100 ng–1 μg gDNA or cDNA Broad Range of Applications: Does not require methylation of DNA or restriction digestion; can be used for bisulfite sequencing Simplified Data Analysis: Higher quality sequence assemblies with short-insert libraries.
文章作者 zzx
上次更新 2015-03-22