
利用biomaRt包下载HGMD公开版的突变位点
文章目录
前面文章介绍了Ensembl的biomart,相信你对biomart应该有所了解了,在此再介绍一种方法,即通过R语言包biomaRt下载HGMD的数据。
HGMD的最新数据是需要购买授权才行,公开版信息不仅滞后,而且不能下载,不能得到基因组位置,在biostart上看到有人说Ensembl整合了HGMD的公开版,心想能获得公开版的数据也不错,于是采用biomaRt包下载。
各位不要高兴,最终的结果是,只得到了所有突变的基因组位置,未能下到具体的突变类型,以及与表型的关系。不过能下载基因组的位置,也算不错,结合对这些位置的注释,能获取不少信息。如果您对这些位置的利用有更多或者更好的想法,欢迎与我讨论。
1,安装biomaRt包
|
|
2,显示ensembl的biomart
|
|
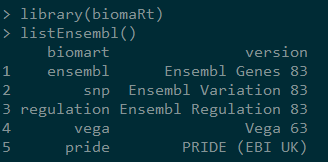
3,选择snp mart
|
|
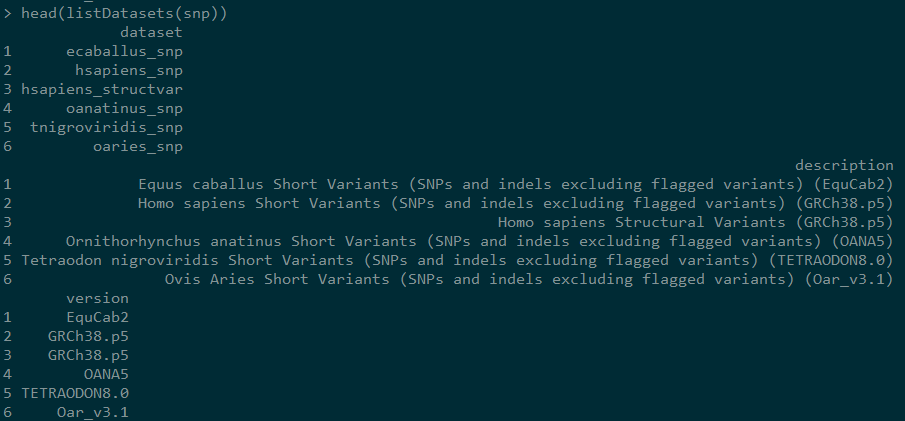
4,显示snp mart的数据集
下述命令会显示snp mart的所有数据集,很明显,我们需要的是hsapiens_snp的数据集,相当于选择哪个物种的数据。description是对数据集的描述,version为版本。
|
|
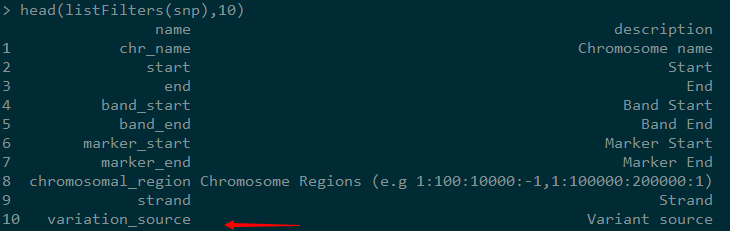
5,显示过滤参数
有时候,我们并不是将整个数据集下载下来,有可能只关注一个基因或者一个RSID,就可以根据显示的过滤参数加上我们的条件,这样得到的数据只是我们关心的。因为我们要获得HGMD的数据,后续要选择variation_source。
|
|
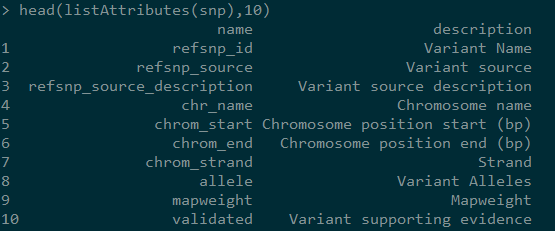
6,显示属性
如果你看过前两篇文章,应该能明白属性其实对应的是我们想得到的关于某一对象的属性,一个属性对应一列,比如SNP的属性就可以包括chr,pos等。后续我选择了’chr_name', ‘chrom_start’,‘chrom_end’,‘refsnp_id’,‘associated_gene’,‘allele’,‘phenotype_description’。我相信,你也关心这些信息,不过最终结果可能没有那么美好,请往下看
listAttributes(snp)
7,通过getBM得到数据
attributes对应你想要获得的属性 filters对应你要过滤的项目,values对应filter的过滤参数 如果你有多个filter,values的参数对应的应该是个list
|
|
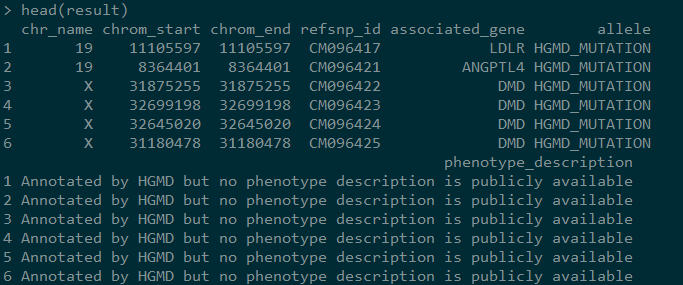
在这里只得到了突变的位置和基因,其他的未公开,想想也对,要是都能下载到了,HGMD的生意也会少了很多。不过HGMD官网没有提供基因组位置,这里能获得,再结合相关基因,这些数据还是很有用的。
Ensembl官网提供的另外两个例子,也可以参考下
|
|
参考:
http://asia.ensembl.org/info/data/biomart/biomart_r_package.html
https://bioconductor.org/packages/release/bioc/html/biomaRt.html
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2016-02-20