
转录组分析新工具流程--HISAT2-stringtie-ballgown
文章目录
一,HISAT2(Hierarchical Indexing for Spliced Alignment of Transcripts2)
HISAT2是一个对比对RNA-seq reads的快速灵敏的spliced alignment工具,HISAT2支持DNA和RNA比对。针对reads覆盖多个外显子,HISAT其包含两种索引:1,global FM索引,代表整个基因组,2,许许多多的local FM索引,每个索引代表~56,000bp,~55,000个local索引覆盖整个基因组。HISAT基于Bowtie2来处理大多数FM索引。心的索引scheme叫做 Hierarchical Graph FM index (HGFM)。HISAT也支持indel和paire end模式,并且支持多线程和SRA Toolkit。HISAT2官网提到HISAT2是HISAT和TopHat2继承,建议大家从HISAT和TopHat2迁移到HISAT2上来。
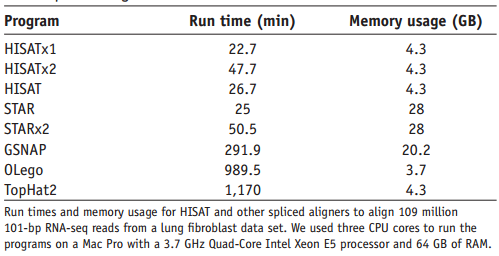
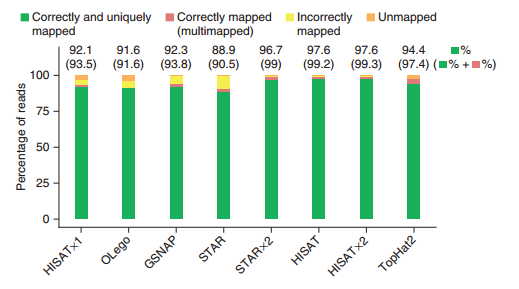
HISAT2的优势如图(图片来自HISAT2文章)
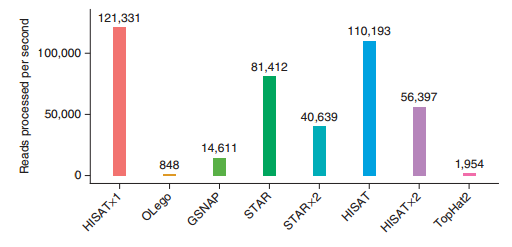 )
)
下载地址: http://ccb.jhu.edu/software/hisat2/downloads/ wget http://ccb.jhu.edu/software/hisat2/downloads/hisat2-2.0.0-beta-Linux_x86_64.zip -P ./ 解 压 缩: unzip hisat2-2.0.0-beta-Linux_x86_64.zip
个人理解,在进行RNA序列比对时,提供剪切位点,能够使软件更快的比对read在基因组的位置。在运行hisat2时,可以通过–known-splicesite-infile指定splice的位置,但HISAT2提到,在build的时候加入splice site更好,所以在build之前,需要准备记录splice sites和exons的文件。HISAT2自带通过基因组gft注释文件提取splice site和exon的脚本。
gtf 文件来自
http://www.gencodegenes.org/releases/19.html#
extract_exons.py hg19.annotation.gtf > exons.txt extract_splice_sites.py hg19.annotation.gtf > splicesites.txt
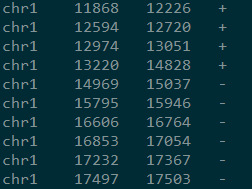
提取后的输出格式为
chr0 based left pos0 based left leftstrand
./hisat2-2.0.0-beta/hisat2-build -f ucsc.hg19.fasta --ss splicesites.txt --exon exons.txt -p 7 ./ucsc.hg19 ## 添加--ss和--exon选项后,需要很大的内存,build 人基因组的话需要200G RAM,如果没有这么大内存,不要添加这两个选项,但要在后续运行hisat时添加 --known-splicesite-infile选项 hisat2-build -f ucsc.hg19.fasta -p 7 ./uscs.hg19 ##大概需要一小时二十分钟 build index的过程和bowtie2很像。 #-q指定fastq格式 hisat2 -q -x ./ucsc.hg19 -1 reads_1.fastq -2 reads_2.fastq -S alns.sam -t ## hisat2 -q -x ./ucsc.hg19 -1 reads_1.fastq -2 reads_2.fastq -S alns.sam --known-splicesite-infile splicesites.txt -t
index时输出的信息
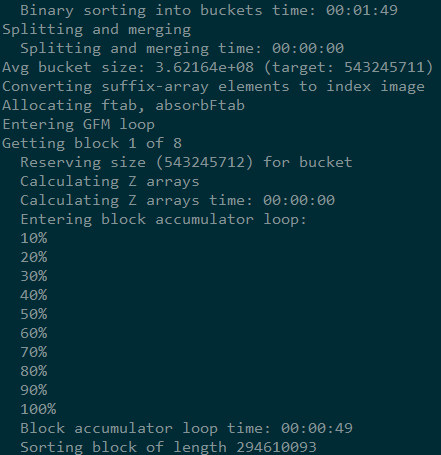
index之后会生成
二,stringtie
比对完之后,就要确定基因或者转录本的表达量了。这里用到了第一步用到的gtf文件。
|
|
运行过程中的输出信息
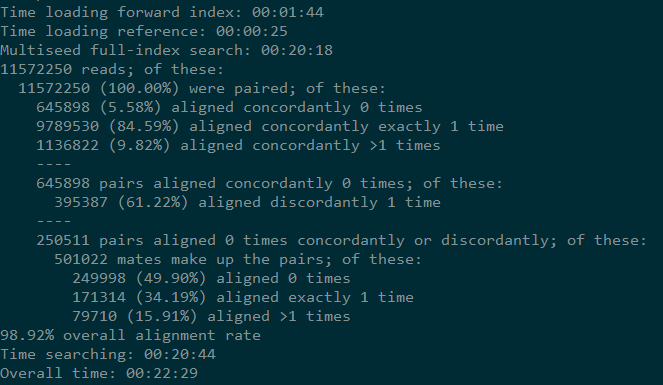{kind=link}
三,ballgown
https://www.bioconductor.org/packages/release/bioc/vignettes/ballgown/inst/doc/ballgown.html
ballgown的流程示例以ballgown自带的数据为例。
source("http://bioconductor.org/biocLite.R")
biocLite("ballgown")
library(ballgown)
data_directory = system.file('extdata', package='ballgown')
#data_directory = c('./stringtie.input/')
bg = ballgown(dataDir=data_directory, samplePattern='sample', meas='all') # load data
# 两组,每组各十个样本
pData(bg) = data.frame(id=sampleNames(bg), group=rep(c(1,0), each=10)) # get pData
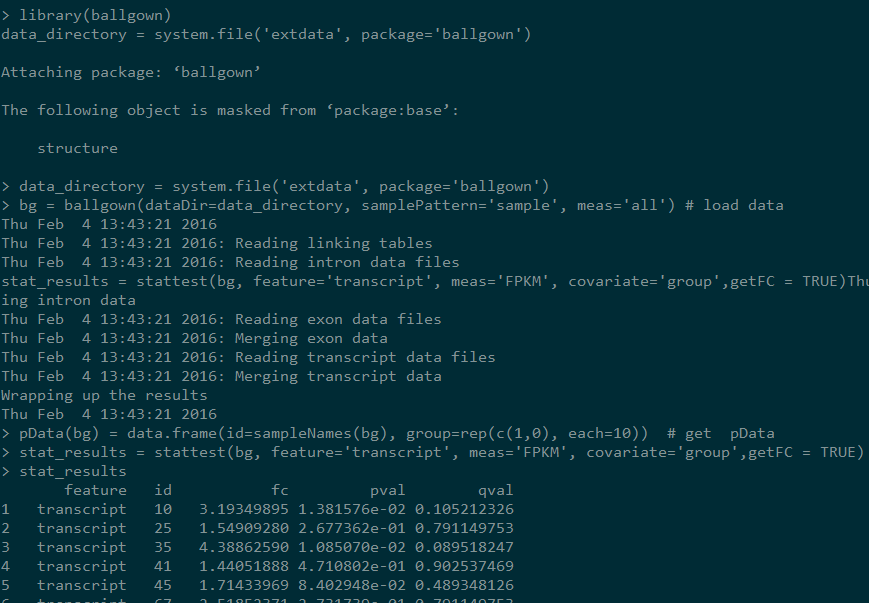
stat_results = stattest(bg, feature='transcript', meas='FPKM', covariate='group',getFC = TRUE)
write.table(stat_results,file="./result.out",sep="t",quote =FALSE,row.names=FALSE)
extdata的文件夹结构如下: extdata/ —sample01/ ——e2t.ctab ——e_data.ctab ——i2t.ctab ——i_data.ctab ——t_data.ctab —sample02/ ——e2t.ctab ——e_data.ctab ——i2t.ctab ——i_data.ctab ——t_data.ctab … —sample20/ ——e2t.ctab ——e_data.ctab ——i2t.ctab ——i_data.ctab ——t_data.ctab
设置getFC为真才会得到fold change的值,输出如下:
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2016-02-16