
三代测序组拼接组装工具Falcon
文章目录
基因组装配工具Falcon工作流程
1 Falcon简介
Falcon (Fast Alignment and CONsensus),是由PacBio(太平洋生物科技公司)新开发的二倍体基因组从头拼接组装工具,由HGAP(Hierarchical Genome Assembly Process)扩展而来,但拥有更快的拼接组装效率。 Falcon的正常运行,需要DAZZ_DB模块用来构建序列的数据库,DALIGNER模块进行序列比对寻找序列之间的重叠和pypeFLOW模块记录和追踪流程进度。
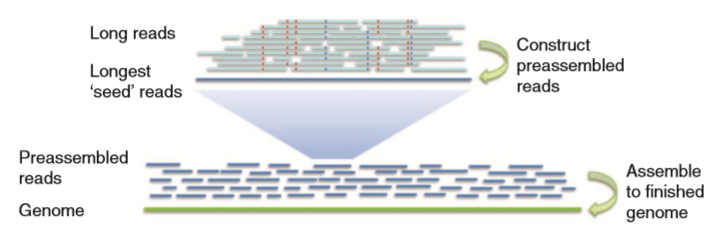
2 Falcon工作步骤
运行:fc_run.py fc_run.cfg fc_run.cfg为配置文件,包含falcon运行所需要的各项参数。
1)构建用于错误修正的原始序列重叠信息
fc_run.py生成prepare.rd脚本,调用Daligner中的fasta2DB生成Dazzler Data Base;调用DB split分割数据块,最终的任务数取决于对序列数据库的分割(-s参数);调用HPCDaligner生成一系列局部比对的命令。具体由生成以rj开头的脚本调用Daligner进行局部比对,生成一系列包含序列之间比对信息的".las"文件。0-rawreads文件夹中的所有.las格式的文件会被以rp开头的脚本调用sort和merge命令合并成一个.las格式的文件。
2)错误修正和预组装
错误修正和预组装的目的在于将原始序列转化为高质量的预组装序列。在以c开头的脚本中,调用LA4Falcon读取las格式文件转换成falcon所能识别的格式,以管道的方式流入fc_consensus.py进行错误修正和预组装,生成consensus序列,放在fasta格式文件中,以用于后续基因组装配。生成的fasta文件个数与以m开头的文件夹个数一致。Fasta文件的头信息可以被Daligner解析。
3)构建错误修正后的序列重叠信息
与第一步相似。参数设置与第一也类似,主要不同在于-e,可以设置较高的两个预组装的序列的关联度,比如-e.96,因为此时序列的错误率已经很低了。
4)过滤重叠
过滤重叠的原因:1),如果一条序列完全包含另外一条序列,这种重叠信息是没价值的;2)序列两端的重叠关系并不需要过多的重叠信息,一定量的重叠信息变可以推算出;3)序列某段的覆盖度较高,可能是由重复区域造成,覆盖度较低,存在较高的错误率等。 确定重叠关系的最大重叠数目,序列两端的最大和最小覆盖度等设置可以在配置文件中的overlap_filtering_setting设置。
5)基于重叠信息构建串图
fc_ovlp_to_graph.py会生成一些用于构建重叠群contig的串图文件。 根据重叠信息,构建图的边,边的信息储存在sg_edges_list文件中,这些边进一步连接成unitigs的信息储存在utg_data文件中。ctg_path则存储根据unitigs构建每个contig的图。fc_ovlp_to_graph.py涉及的参数有: –min_len MIN_LEN:用于装配拼接的序列最低长度 –min_idt MIN_IDT:用于装配拼接的序列之间的最低比对相似度 –lfc:解决串图中的节点时,使用local flow constraint方法而不是最佳重叠策略。
6)基于图构建重叠群(contig)
fc_graph_to_contig.py根据图路径和序列,构建contig。
3 Falcon安装
目前PacBio提供了整个falcon依赖模块的integrate版本,详 见 https://github.com/PacificBiosciences/FALCON-integrate
只需按照步骤执行
git clone git://github.com/PacificBiosciences/FALCON-integrate.git cd FALCON-integrate make init make virtualenv make check make -j install make test # to run a simple one
但前提是您的电脑必须联网,如果您的电脑没有联网,可以下载Falcon20150728,执行install-offline.sh。
4 Falcon 配置文件fc_run.cfg
[General] # list of files of the initial bas.h5 files input_fofn = input.fofn 指出所有输入数据 #input_fofn = preads.fofn input_type = raw 标明序列类型,即是否已经完成了错误修正 #input_type = preads # The length cutoff used for seed reads used for initial mapping length_cutoff = 10000 用于错误修正的种子序列的最低长度 # The length cutoff used for seed reads usef for pre-assembly length_cutoff_pr = 10000用于构建重叠的预组装种子序列的最低长度 # target = pre-assembly # target = mapping target = assembly 用于控制Daligner任务队列,-pe指定并行环境,-q指定要提交到的队列,8表示线程。 sge_option_da = -pe smp 8 -q bigmem sge_option_la = -pe smp 2 -q bigmem sge_option_pda = -pe smp 8 -q bigmem sge_option_pla = -pe smp 2 -q bigmem sge_option_fc = -pe smp 24 -q bigmem sge_option_cns = -pe smp 8 -q bigmem pa_concurrent_jobs = 32 fc_run.py提交并发任务的数目 cns_concurrent_jobs = 32 ovlp_concurrent_jobs = 32 -dal决定单个任务中相互比对的数据块的数目(影响生成的任务数),-e序列之间的关联程度,-s trace points sparse ,-l 低于-l的序列将被忽略,-t Tuple suppression frequency?????,-h Hit threshold (in bp.s) pa_HPCdaligner_option = -v -dal128 -t16 -e.70 -l1000 -s1000 ovlp_HPCdaligner_option = -v -dal128 -t32 -h60 -e.96 -l500 -s1000 -s序列数据库分割后,每个数据块的Mb大小,低于-x阈值长度的序列将被忽略 pa_DBsplit_option = -x500 -s400 ovlp_DBsplit_option = -x500 -s400 --min_cov种子序列的最低覆盖度;--max_n_read用于错误修正的最大序列数目,防止重复区域中序列的影响,--output_multi output multi correct regions,--min-idt minimum identity of the alignments used for correction,--max_n_read 用于生成consensus的最低序列数目,--n_core生成consensus的线程数 falcon_sense_option = --output_multi --min_idt 0.70 --min_cov 4 --local_match_count_threshold 2 --max_n_read 200 --n_core 6 --max_diff序列两端的最大覆盖度差异,max_cov序列两端的最大覆盖度--min_cov最低覆盖度,bestn如有输出指定数目的最好的重叠 overlap_filtering_setting = --max_diff 100 --max_cov 100 --min_cov 1 --bestn 10
参考:
https://github.com/PacificBiosciences/FALCON/wiki/Manual
https://github.com/PacificBiosciences/FALCON-integrate
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2015-07-28