
用exceRpt定量miRNA、piRNA、tRNA
文章目录
exceRpt:The extra-cellular RNA processing toolkit
关于miRNA的定量工具其实有很多,有人用STAR或者bowtie比对,然后自己定量。我喜欢打包的解决方案,避免造轮子,我经常用的是miRDeep2。朋友问piRNAR如何定量,是否可以用exceRpt。于是和他一起研究了下exceRpt,发现exceRpt对miRNA的定量基本上与miRDeep2的结果一致,感觉exceRpt靠谱。
exceRpt目地址:http://github.gersteinlab.org/exceRpt/
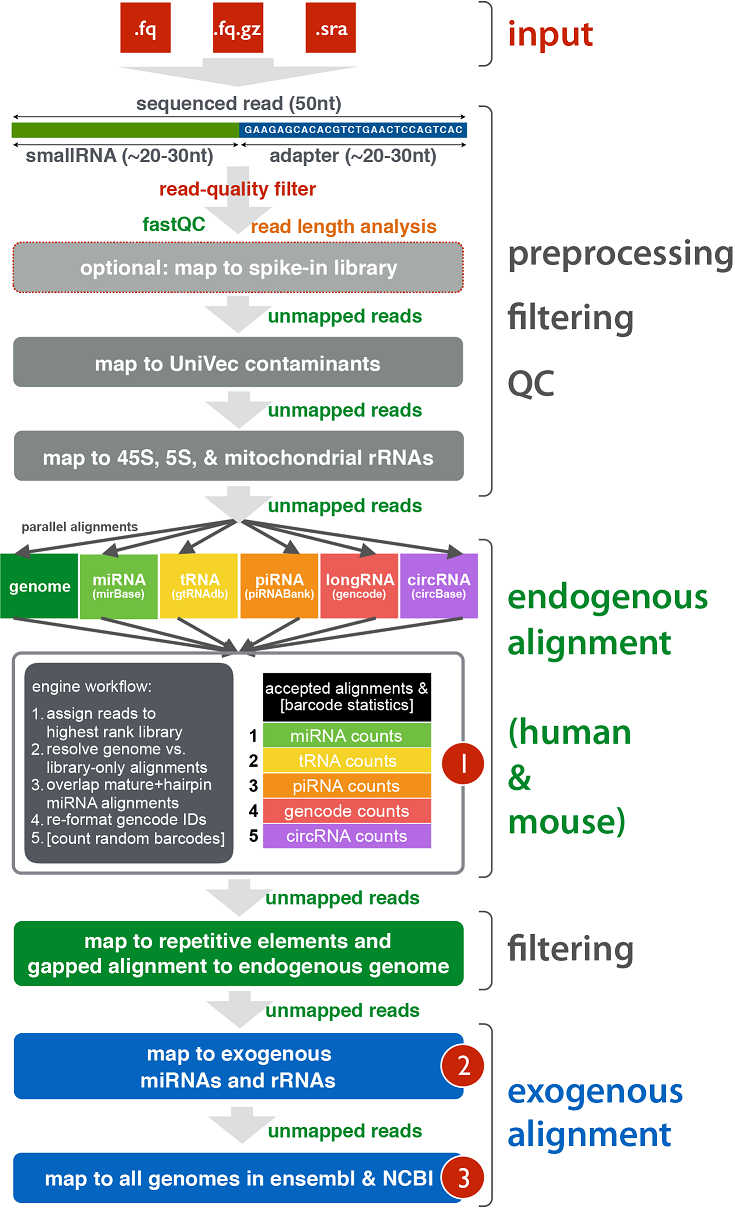
主要思想是先去掉无关的reads,比如45S, 5S,rRNA等,然后同时比对genome, miRNA(mirBase),tRNA( gtRNAdb), piRNA( piRNABank),longRNA(gencode), circRNA(circBase),看reads和哪种类型的的RNA最接近,然后定量。
Tutorial
安装
exceRpt用的是docker
|
|
下载对应的数据库
|
|
运行
|
|
docker -v的命令是把本地文件夹挂载到docker上,
/datadir就是fastq文件所在的文件夹
hg38文件夹就是下载的数据库文件,一定要挂载到docker上的/exceRpt_DB/下,
ADAPTER_SEQ中指定小RNA建库时的接头序列
如果遇到adapter两端还有随机序列,可以通过TRIM_N_BASES_5p或者TRIM_N_BASES_3p指定左右两端需要切去多长的碱基,RANDOM_BARCODE_LENGTH则是两端同时截取。
后续处理
用项目提供的R脚本,则可以获得count per million的数据。(可能会安装很多包)
|
|
execRpt和miRDeep2在miRNA定量上的相关性
我们用了自己的一个样本进行了测试。
miRDeep2在定量的时候,来自不同precursor的mature会被认为两个,后续分析一般会合并,最后有近2700个miRNA,其中312个有表达的。execRpt则仅输出比对上的,有314个,而且execRpt会把家族中相近的当作一个,比如hsa-miR-151a-5p|hsa-miR-151b在execRpt中是一个记录,当作一个miRNA,而miRDeep2则会输出两条记录,当作两个miRNA,这也是execRpt和miRDeep2不同的地方。
最终有289个共同被检测到的miRNA,基本一致,并且execRpt和miRDeep2从绝对定量上来看差异也非常小,我们根据miRNA的排了序,结果如图。
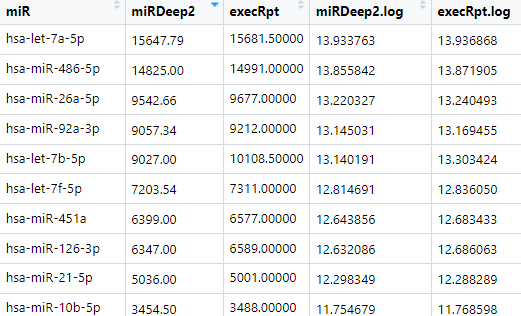
我们画了散点图,来看相关性,基本上就是一条直线,只有个别的miRNA定量有差异。

如果miRNA定量没有问题的话,我们可以推荐piRNA,tRNA的定量效果应该还不错。能一步同时对多种RNA进行定量,还不用自己造轮子,👍。
其他工具
其他用于非编码小RNA分析的工具还有,但我没有测试:
COMPSRA(后期维护不好,文档显得有点乱): a COMprehensive Platform for Small RNA-seq data AnalySis:https://github.com/cougarlj/COMPSRA
seqpac:A Framework for small RNA analysis in R using Sequence-Based Counts:https://rpubs.com/signeskog/seqpac
SortMeRNA:https://github.com/sortmerna/sortmerna,可以指定多种reference,比如鉴定病毒序列等
bcbio-nextgen(软件难安装):https://bcbio-nextgen.readthedocs.io/en/latest/contents/small_rnaseq.html
这篇文章有讲自己如何比对non-coding RNA:https://www.cell.com/molecular-therapy-family/nucleic-acids/pdfExtended/S2162-2531(18)30210-5
https://github.com/bcbio/bcbio-nextgen/issues/2439 这个里面提到了piRNA等注释文件
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: 感谢Caiming提供的测试数据&Jason
#####################################################################
文章作者 zzx
上次更新 2022-05-24