
可变多聚腺苷酸化Alternative Polyadenylation (APA) 检测
文章目录
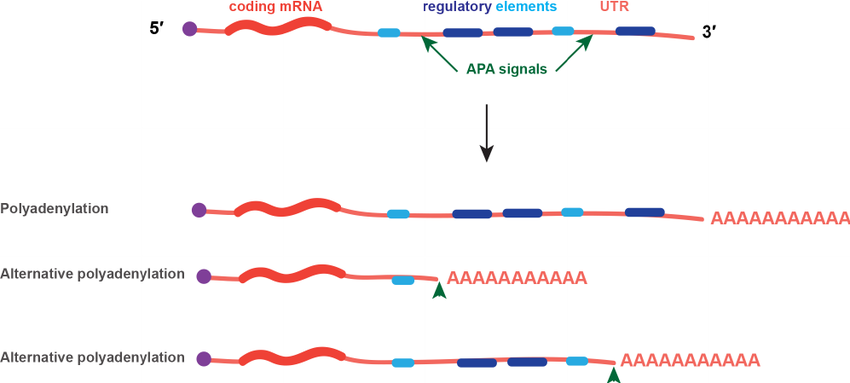
可变多聚腺苷酸化Alternative Polyadenylation (APA),如下图所示(图片来自参考),在不同的APA信号位点切割,然后添加polyA。这种调控机制属于转录后调控,可能会影响蛋白的序列(发生在编码区),也可能影响蛋白的稳定性(比如非编码区内的miRNA的调控区域)。其实也是可变剪接的一种情况。
常用的软件是Dapars,这个软件现在也有了升级的版本Dapars2。参考: https://github.com/ZhengXia/dapars https://github.com/3UTR/DaPars2 分析流程很相似,Dapars2多了 normalize library sizes 。
第一步:生成Wiggle文件
这一步其实是统计每个点或者区域的覆盖度,便于后续计算,用bedtools统计STAR比对后的结果即可。
genomeCoverageBed -bg -ibam Aligned.sortedByCoord.out.bam -split > Aligned.sortedByCoord.out.wig
第二步: Generate region annotation: DaPars_Extract_Anno.py
这一步需要从bed12格式的文件中,提取远端ployA的位点,进而便于推测近端polyA的位点。 1)作者推荐从UCSC上下载,http://genome.ucsc.edu/cgi-bin/hgTables?command=start,选择bed格式之后,选这whole gene即可。 2)如果你有特定的gtf文件,可以将gtf文件转成bed12格式,先将gtf转成genPred格式,然后再转成bed12格式。http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/上有现成的工具
第三步(Dapars2): Generate mapped reads files for all Samples
这一步是为了 normalize library sizes ,统计每个样本比对上的reads数据,格式如下,与下一步的顺序一致。Dapars2才有这一步。
第四步: Run DaPars
配置文件,运行即可。结果中会告诉ΔDPUI也就是远端APA位点的利用率的差值及显著性,根据这些挑选候选。然后回到IGV上进行查看,如下基因方向从右到左,可以看出来前六个和后三个样本在最后一个外显子上的覆盖度截然不同,不同的地方可能就是发生了APA事件,然后添加polyA。

参考
Weil T T. Post-transcriptional regulation of early embryogenesis[J]. F1000prime reports, 2015, 7. https://leilisysbio.github.io/DaPars2_Documentation/DaPars2.html
https://www.jianshu.com/p/21b697cec428
##################################################################### #版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任 #Author: Jason
####################################################################
文章作者 zzx
上次更新 2020-02-03