
什么是ubam文件,为什么ubam文件比fastq文件好
文章目录
ubam是Unmapped BAM Format,是BAM文件的一个变种,里面的read是未经map的。大部分测序供应FASTQ文件,这是最常见的测序分析的起始文件。FASTQ文件的优势是,压缩比比bam文件好,解压速度快。
但与ubam文件相比,FASTQ并不是最理想的:
1)单个文件更容易分析
在双端测序中,有些软件希望配对的reads放在一个文件中,有些希望配对的文件,有些软件直接根据read在文件中的位置判断read是否配对,当然现在FASTQ通过在文件中加入/1和/2来表明配对的read解决这一问题。在生信分析的时候,一个FASTQ文件往往和另一个FASTQ文件关联配对,比如R1和R2,往往会花费更多时间来验证read是否配对。但如果通过bam文件的话,会更简单。只需要在FLAG这个地方加入对应的值,比如77和141,就能指定。单个文件更加简单,能储存更多metadata的信息。
2)BAM文件可以储存更多信息
实际上,正是这个原因,我才开始用ubam文件的。 因为FASTQ文件格式的问题,很多其他信息不能存在FASTQ文件中,而BAM/SAM文件可以储存很多信息,只需加tag或者header就行。如果你想放,可以放用了什么软件,平台,样本标签等。

上图是不利用ubam文件的流程,下图是利用ubam文件的流程。
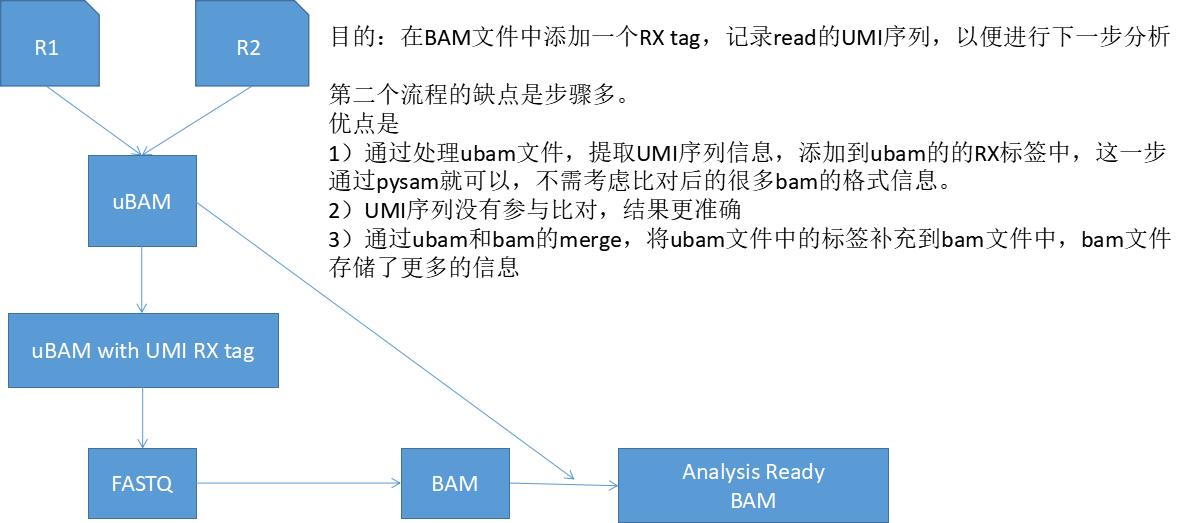
这个我体会很深,比如我在分析带分子标签UMI的数据时,需要在bam文件中添加read的UMI标签序列,具有相同的UMI标签的序列说明来自同一个分子。如果我将带UMI标签的reads直接比对基因组,UMI标签序列会影响比对质量,而且后续对bam文件的处理提取UMI时,需要考虑CIGAR标签,这个时候就头大了。实际上,我是先将fastq转成ubam文件(picard的FastqToSam工具),然后提取ubam文件中的UMI,生成新的ubam,将ubam转成fastq文件(reads都放在一个文件fastq文件中,bwa能识别其中的配对reads),与参考基因组进行比对。这个时候问题就来了,比对之后生成的bam文件,不包含我提取的UMI标签序列信息,因为这个信息在ubam文件中。不要着急,picard的另外一个工具MergeBamAlignment,可以将ubam文件和bam文件合并,这样ubam文件中的tag信息,比如UMI标签信息,就会补充到新的bam文件中。这样生成的bam文件供下一步数据分析。
3)BAM能实现随机读写
BAM文件采用的是BGZF压缩,可以通过随机读写快速读取文件特定位置的数据。对于未比对的reads(unaligned reads),这个功能不是很吸引人,因为后续序列比对都是顺序比对的,不可能跳着比对。
参考: https://blastedbio.blogspot.com/2011/10/fastq-must-die-long-live-sambam.html
####################################################################
#版权所有 转载请告知 版权归作者所有 如有侵权 一经发现 必将追究其法律责任
#Author: Jason
####################################################################
文章作者 zzx
上次更新 2018-07-12